










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban ene. 2014
RESEARCH ARTICLE
Selective Adsorption of Tetrahydropalmatine by a Molecularly Imprinted Polymer with Modified Rosin Cross-linker
Pengfei LiI, II; Caihua MuI; Ting WangI, II; Fuhou LeiI, II, *; Hao LiI; Qin HuangI, II; Juying ZhouI, II
ISchool of Chemistry and Chemical Engineering, Guangxi University for Nationalities, Nanning 530006, P.R. China
IIGuangxi Key Laboratory of Chemistry and Engineering of Forest Products, Nanning 530006, P.R. China
ABSTRACT
A molecularly imprinted polymer (MIP) containing a phenanthrene skeleton was prepared by suspension polymerization with ethylene glycol maleic rosinate acrylate (EGMRA) as the cross-linker, tetrahydropalmatine (THP) as the template, and methacrylic acid as the functional monomer. A non-imprinted polymer (NIP) was similarly prepared and treated, but in the absence of THP The MIP and NIP were characterized by scanning electron microscopy and nitrogen sorption and thermal gravimetric analyses. The time taken by the MIP to reach THP (C0 = 2.8 mmol L1) adsorption equilibrium was 4.5 h, and the recognition factor of the MIP for THP was 2.09. The change in microcalorimetric heat flow during adsorption revealed that the MIP had a higher affinity to THP compared with NIP. Selective adsorption experiments demonstrated the high affinity and THP selectivity of the MIP.
Keywords: Molecularly imprinted polymer, tetrahydropalmatine, selective adsorption, microcalorimetry, modified rosin.
1. Introduction
Molecular imprinting is a technique for preparing molecular imprinted polymers (MIP) that contain a large number of cavities. Cavities are complementary in shape, size and functionality to a specific target molecule, and can recognize and bind the target molecule with high selectivity.1,2 MIPs are straightforward and inexpensive to prepare, and are relatively stable.3,4 They are used in sample pretreatment,5,6 sensors,7,8 chromatography9"12 and catalysts.13,14
MIPs are usually prepared using ethylene glycol dimethacrylate (EGDMA) as a cross-linker,15 but such MIPs generally exhibit poor rigidity. The irreversible distortion of the molecularly imprinted holes adversely affects recognition performance and selectivity. Considerable effort has been focused on improving the mechanical strength and chemical stability of MIPs. Developing alternative cross-linkers is likely to facilitate the synthesis of MIPs with high selectivity and stability.
In this study, the cross-linker ethylene glycol maleic rosinate acrylate (EGMRA) (Fig. 1) was synthesized from rosin,16 and was used to prepare a MIP EGMRA was used as a monomer for affinity separation by introducing the phenanthrene skeleton into the polymer to improve its mechanical strength and stability. It overcomes the above-discussed disadvantages of conventional molecular imprinting technology.
Tetrahydropalmatine (THP) is an alkaloid obtained from rhizoma corydalis. THP is a traditional Chinese herbal remedy used as a sedative, antispasmodic, analgesic and decongestant, and is a pharmaceutical product in the Peoples Republic of China.17 THP is of interest because it is a sedative with low toxic-ity and addictive properties and so its separation and purification is important. There are currently no reports of using MIPs prepared from EGMRA for the separation of THP.
In this study, a MIP was prepared by suspension polymerization using EGMRA as the cross-linker, THP as the template and methacrylic acid (MAA) as the functional monomer. The obtained particles were characterized by scanning electron microscopy (SEM), nitrogen sorption analysis with Brunauer-Emmett-Teller theory (N2-BET) and thermogravimetric analysis (TGA). The adsorption properties of the MIP were investigated by equilibrium adsorption experiments. The quantity of heat adsorbed was determined via microcalorimetry. The selectivity was elucidated from the different binding abilities of THP and theo- phylline (TP).
2. Experimental
2.1 Preparation of THP-imprinted Polymers
THP (1.4 mmol), EGMRA (14 mmol) and MAA (42 mmol) were dissolved in 60 mL of ethyl acetate by sonication. 0.36 g of azodiisobutyronitrile (AIBN) and 4.5 mL of porogen were added, and the solution was sonicated to form an organic phase. The organic phase was added to aqueous sodium dodecyl sulfate (0.5 mg mL-1, 400 mL) under constant stirring, and the mixture was thermally polymerized at 78 °C for 20 min, and then at 95 °C for 40 min. The polymer was thoroughly washed with acetone to remove any unreacted monomer, and was then thoroughly washed with water until no THP was detected in the effluent. A non-imprinted polymer (NIP) was similarly prepared and treated, but in the absence of THP
2.2. Template Removal
After the imprinted polymer was prepared, the THP template needed to be removed, to leave vacant cavities complementary in shape, size and functionality. Methanol/acetic acid (80:20 v/v) extraction for 48 h was found to be suitable for quantitatively removing the THP template. Extraction was repeated until negligible THP was detected in the washing solution, as shown by the absorbance at 280 nm, (Shimadzu UV1800, China).
2.3. Characterization of Materials
Scanning electron microscopy: the morphologies of the obtained polymers were determined via SEM (Hitachi S-3400N, Japan). Prior to visualization, a small section of the polymer was placed on the SEM sample holder and was then sputter-coated with gold.
Nitrogen adsorption desorption isotherms were collected on a Micromeritics ASAP2020M instrument at 77 K after the sample had been degassed in a flow of N2 at 373 K overnight. The Brunauer-Emmett-Teller (BET) method was utilized to calculate the specific surface area; pore volume and pore diameter distribution was derived from the adsorption isotherms by the Barrett-Joyner-Halenda (BJH) model.
Thermogravimetric analysis (TGA) was performed on chitosan and modified chitosan by using a NETZSCH STA-449F3 Jupiter instrument. Experiments were performed with 4-5 mg of the sample under a dynamic nitrogen atmosphere flowing at a rate of 50 mL min-1 and at a heating rate of 10 K min-1.
2.4. Adsorption Experiments
0.25 g of MIP (or NIP) was placed in a 100 mL conical flask, with a known concentration of THP (defined as C0)in50mLof ethanoVwater (50:50 v/v). The flask was agitated at 30 °C for 4.5 h, and the solution centrifuged. The THP concentration of the solution was determined by measuring the absorbance at 280 nm, using a spectrophotometer. The sorption quantity Qe (mmol g-1) was calculated from:18

where Co and Ce are the initial and equilibrium THP concentrations, respectively (mmolL-1), Vis the THP solution volume (mL) and m is the MIP (or blank sample) weight (g).
2.5. Specific Adsorption Experiments
2.0 mmol L-1 solutions of THP and TP were prepared. The Qe of the MIP and NIP for these three analytes was obtained by the method in section 2.3.
The specific recognition properties of the MIP were evaluated with the distribution coefficient (K) and recognition factor (ö). K was calculated from:19

where the units of K are L g - 1. θ was calculated from:

where Kmip and Knip are the distribution coefficients of the MIP and NIP, respectively.
2.6. Selectivity Adsorption Experiments
The selectivity of the MIP was evaluated from competitive binding studies, using a solution containing 2 mmol L-1 of THP and TP, at optimum adsorption conditions. The THP and TP concentrations remaining in solution were determined by high performance liquid chromatography (HPLC, Shimadzu LC-15C, China). Prior to analysis, the supernatant was filtered through a 0.45 µm membrane. The HPLC conditions were: mobile phase, methanol; flow velocity, 1.0 mL min-1; wavelength, 280 nm; column temperature, 30 °C; sample volume, 20 µL.
2.7. Microcalorimetry
The energy evolution of adsorption was measured by a Setaram C80 microcalorimeter with a mixing cell. Energy changes in the adsorption of THP were detected at 298.15 K. The THP concentration and volume were 2.0 mmol L-1 and 1.0 mL, respectively. The mass of the MIP (or NIP) was 0.25 g.
3. Results and Discussion
3.1. Polymer Characterization
SEM images of the MIP and NIP are shown in Fig. 2a and 2b, respectively. The images reveal the loose structure of the MIP particles, and the numerous cavities and channels present on the particle surface. By contrast, the structure of NIP was more compact. This difference probably resulted from the different template and porogen molecules used in the polymerization.20
The textural properties of the MIP and NIP are given in Table 1. The MIP exhibited a much smaller surface area and pore volume than the NIP. The average pore diameters were > 50 nm, so the MIP and NIP structures were predominantly constituted of macropores.

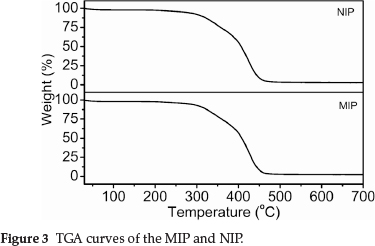
The TGA results in Fig. 3 show that the TGA curves of MIP and NIP were not significantly different. The addition of the THP template did not appear to significantly affect the thermal properties of the MIP. The principal chains of the cross-linked MIP (or NIP) began to degrade at ~280 °C, and the final decomposition temperature was ~470 °C.
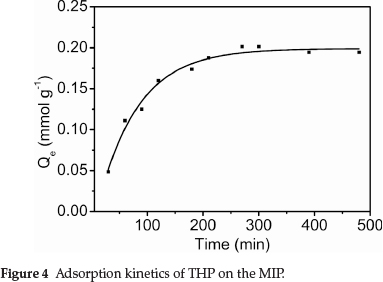
3.2. Adsorption Kinetics of THP on the MIP
Figure 4 shows the dynamic curve for THP (C0 = 2.8 mmol L-1) adsorption on the MIP particles. Adsorption capacity increased with increasing contact time. The adsorption rate was initially very fast, with about 50 % of adsorption having been reached within 60 min. The adsorption capacity subsequently increased more slowly with increasing contact time, reaching a plateau at 4.5 h. THP preferentially adsorbed on the MIP surface sites, and adsorption was faster at the beginning. Increasing adsorption of THP on the MIP surface decreased the binding sites available for further adsorption. Consequently, the adsorption rate decreased with increasing surface coverage.
3.3. Adsorption Isotherms
The binding characteristics of the MIP were determined via the equilibrium adsorption from fixed THP concentration solutions. The adsorption isotherm on the MIP was determined at THP concentrations from 1.3 to 4.2 mmol L-1. The results are shown in Fig. 5.
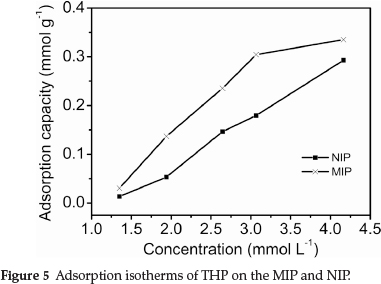
Figure 5 shows that the adsorption capacity of the MIP and NIP increased with increasing THP concentration. The amount of adsorbed THP on the MIP was much larger than that of the NIP at the same THP concentration. This indicated that the MIP was capable of specifically adsorbing the template, because the imprinted cavities are complementary to the template in size, shape and spatial arrangement of functional groups. However, the NIP could not form specific recognition sites in the absence of the template.21
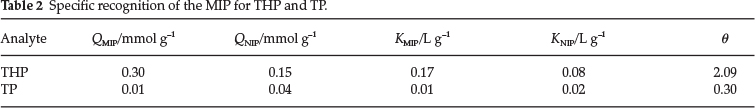
3.4. Specific Recognition of the MIP
The competitive adsorption of the MIP for THP with respect to TP was carried out, as described in section 2.3. The results are shown in Table 2. d was found to obey the trend θTHP > θTP. The adsorption quantity and distribution coefficient of the MIP for THP were much larger than those for TP. The MIP exhibited a high affinity to the template, because the polymers have cavities of complementary size, shape, and functionality to the template. TP is smaller than the template, so its adsorption by the MIP is nonspecific.
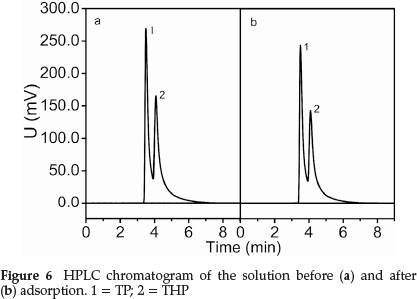
3.5 Selectivity of the MIP
Figure 6 shows that the adsorption peak area at pre- and post-adsorption changed significantly. The TP content decreased by 15.7 %, and the THP content decreased by 8.1 %. This indicated the high selectivity of the MIP for THP in the presence of TP.
3.6. Microcalorimetry
Microcalorimetric heat flow curves for adsorption by the MIP and NIP are shown in Fig. 7. The initial thermal disturbance was caused by the loading of the stainless steel sample cells. The subsequent flat region (t1 = 0-830 s) indicated negligible thermal events. The curve during this period was taken as the baseline for the subsequent thermal peaks.
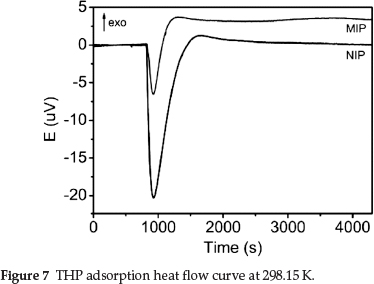
All physical and chemical processes are accompanied by heat exchange. Strong endothermic peaks and relatively weak exothermic peaks were observed in the two curves. The adsorption of the MIP and NIP was endothermic and exothermic, respectively, with a ∆HMIP = 4.4675 J and ∆HNIP = 4.9693 J. When the polymers and solutions were mixed, adsorption began and the total energy rapidly decreased, with subsequent endother-mic peaks. The endothermic peak area of the NIP was much larger than that of the MIP. With increasing adsorption, the endothermic event gradually changed into an exothermic event, and exothermic peaks appeared. The heat flow curves eventually stabilized when adsorption equilibrium was reached. The heat flow curves indicated that the adsorption of THP by the MIP and NIP was of a different nature, and that the MIP more easily adsorbed THP than the NIP.
4. Conclusions
A MIP that targeted THP was prepared through the polymerization of an EGMRA cross-linker, THP template and MAA functional monomer. The MIP exhibited a loose, porous structure and high thermal stability. The adsorption of THP by the MIP required 4.5 h to reach equilibrium at the initial THP concentration was 2.8 mmol L-1, and the recognition factor of the MIP was 2.09. The MIP could be used to separate THP extracts in pharmacology applications.
Acknowledgements
The authors thank the National Natural Science Foundation of China (Nos. 31360162, 51203027), the Guangxi New Century Hundred, Thousand and Ten Thousand Talent Project (No. 201292), the Natural Science Foundation of Guangxi University for Nationalities (Nos. 2013MDYB029, 2013MDQN038), and the Guangxi Natural Science Foundation of China (No. 2012GXNSFAA053031) for financial support.
References
1 B.T.S. Bui and K. Haupt, Anal. Bioanal. Chem., 2010,398, 2481-2492. [ Links ]
2 M. Zayats, M. Kanwar, M. Ostermeier and P.C. Searson, Macro-molecules., 2011,44, 3966-3972. [ Links ]
3 R.V. Vitor, M.C.G. Martins, E.C. Figueiredo and I. Martins, Anal. Bioanal. Chem, 2011,400, 2109-2117. [ Links ]
4 L.J. Schwarz, B. Danylec, Y.Z. Yang, S.J. Harris, R.I. Boysen and M.T.W Hearn, J. Agric. Food. Chem, 2011, 59, 3539-3543. [ Links ]
5 P.P. Qi, J.C. Wang, J. Jin, F. Su and J.P. Chen, Talanta., 2010, 81, 1630-1635. [ Links ]
6 Q.J. Zhu, L.P. Wang, S.F. Wu, W. Joseph, X.H. Gu and J. Tang, Food. Chem., 2009, 113, 608-615. [ Links ]
7 O.Y.F. Henry, D.C. Cullen and S.A. Piletsky, Anal Bioanal Chem., 2005, 382, 947-956. [ Links ]
8 L. Liu, X.C. Tan, X.X. Fang, Y.X. Sun, F.H. Lei and Z.Y. Huang, Electroanalysis, 2012,24, 1647-1654. [ Links ]
9 J.P. Fan, L. Zhang, X.H. Zhang, J.Z. Huang, S. Tong, T. Kong, Z.Y. Tian and J.H. Zhu, Anal. Bioanal. Chem., 2012,402, 1337-1346. [ Links ]
10 B. Boyd, H. Bjork, J. Billing, O. Shimelis, S. Axelsson, M. Leonora, E. Yilmaz, J. Chromatogr. A., 2007,1174, 63-71. [ Links ]
11 H. Khan, T. Khan and J.K. Park, Sep. Purif. Technol.,2008,62,363-369. [ Links ]
12 W.J. Cheong, S.H. Yang and F. Ali, J. Sep. Sci., 2013,36, 609-628. [ Links ]
13. S. Muratsugu and M. Tada, Acc. Chem. Res., 2013,46, 300-311. [ Links ]
14 M. Resmini, Anal. Bioanal. Chem., 2012,402, 3021-3026. [ Links ]
15 H. Kim, K. Kaczmarski and G. Guiochon, Chem. Eng. Sci., 2005, 60, 5245-5444. [ Links ]
16 J.S. Lee and S.L. Hong, Eur. Polym. J., 2002,38, 387-392. [ Links ]
17 D.G. Barceloux, Medical toxicology of natural substances, in Foods, Fungi, Medicinal Herbs, Plants, and Venomous Animals, John Wiley & Sons, USA, 2008, pp. 518-521. [ Links ]
18 Y.F. Kang, W.P. Duan, Y. Li, J.X. Kang and J. Xie. Carbohyd. Polym., 2012, 88. 459-464. [ Links ]
19 B.J. Gao, J. Wang, F.Q. An and Q. Liu, Polymer., 2008,49, 1230-1238. [ Links ]
20 Y.J. Yang, J.Y. Li, Y.R. Liu. J.Y. Zhang. B. Li and X.P. Cai, Anal. Bioanal. Chem., 2011,400, 3665-3674. [ Links ]
21 Z.H. Zhang, X. Yang, X. Chen, M.L. Zhang, L.J. Luo, M.J. Peng and S.Z. Yao, Anal. Bioanal. Chem., 2011,401, 2855-2863. [ Links ]
Received 28 November 2013
Revised 6 March 2014
Accepted 10 March 2014
* Author for correspondence. E-mail: leifuhou@gmail.com


{kind=link}
{kind=link}
{kind=link}