










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
SHORT COMMUNICATION
1H NMR-based kinetic-mechanistic study of the intramolecular trans-esterification of 2-exo-3-exo-dihydroxybornane monoacrylate esters
Andrew R. Duggan; Lulama P. Mciteka; Kevin A. Lobb; Perry T. Kaye*
Department of Chemistry, Rhodes University, Grahamstown, South Africa
ABSTRACT
A 1H NMR study of the acid-catalyzed, intramolecular trans-esterification between isomeric 2-exo-3-exo-dihydroxybornane monoacrylate esters has afforded insights into the reaction mechanism and permitted the determination of kinetic and thermodynamic parameters for the pseudo-first-order processes.
Keywords: Trans-esterification, kinetics, dihydroxybornane monoacrylate esters, 1H NMR studies.
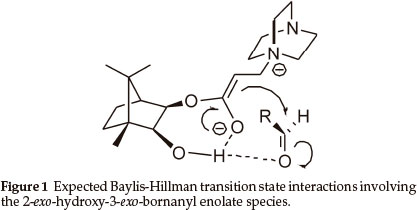
The Baylis-Hillman reaction has attracted considerable attention1 and, in our own laboratories, has been used to construct various benzannulated heterocyclic systems.2 A number of asymmetric Baylis-Hillman syntheses have been reported3 and, as part of an ongoing asymmetric synthesis programme,4 we decided to explore the possibility of using 2-exo-3-exo-dihydroxybornane monoacrylate esters as chiral Baylis-Hillman substrates. It was anticipated that the title compounds would provide: i) a rigid scaffold to minimize conformational flexibility; ii) a proximate asymmetric moiety to induce diastereofacial selectivity; and iii) a hydroxyl group to increase the reaction rate5 and enhance transition state rigidity via intramolecular hydrogen bonding, as illustrated in Fig. 1.
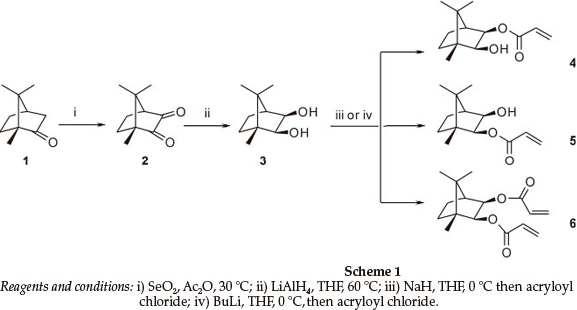
The synthesis of the title compounds is outlined in Scheme 1. Thus, SeO2 oxidation of camphor 1 afforded camphorquinone 2 (84 %),6 LAH reduction of which gave 2-exo-3-exo-dihydroxy-bornane 3 in excellent yield (96 %). Treatment of the dihydroxybornane 3 with BuLi, followed by acryloyl chloride at 0 °C, afforded a mixture of mono- and diester products (4: 34 %; 5:47.8 %; and 6:10.8 %). However, while chromatographic separation of the products could be achieved, the tendency of either monoester to undergo equilibration (see Fig. 2) in CDCl3 soon became apparent, prompting a detailed investigation of the kinetics of this unexpected, and apparently spontaneous, transesterification.
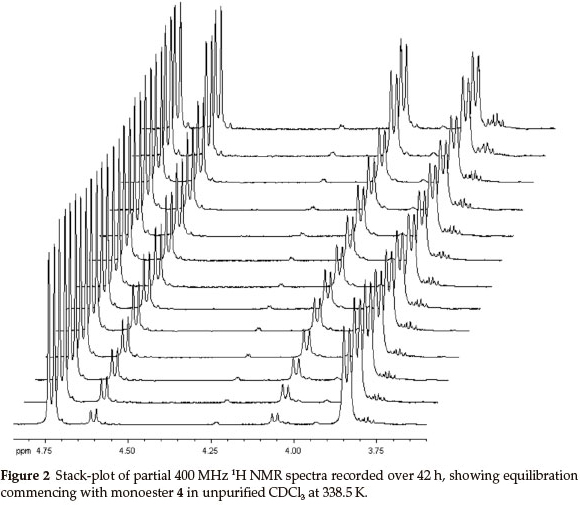
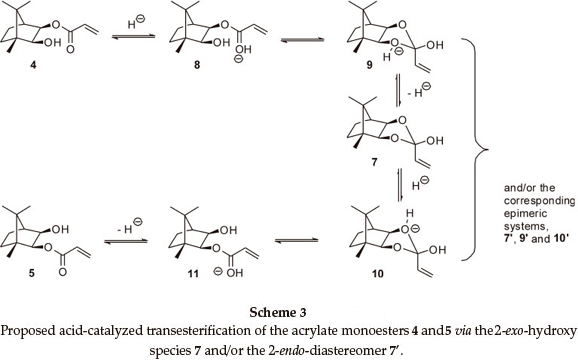
At 400 MHz, the 1H NMR 2-H and 3-H signals for both monoesters are well resolved and proved to be excellent probes to monitor the trans-esterification (Fig. 2). In order to establish the final equilibrium concentrations of the isomeric esters, the NMR tube, containing the mixture was kept in a constant temperature (323 K) water bath and further 1H NMR spectra were recorded at regular intervals until the relative concentrations remained essentially unchanged for ca. 12 h. While the possible intermediacy of transient species such as the 2-exo-hydroxy species 7 (and/or the 2-endo-diastereomer 7'; Scheme 2) was recognized, there was no experimental evidence for their existence.
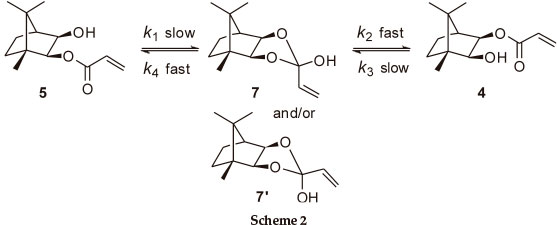
Consequently, it may be assumed that (k1 << k2; Scheme 2) and (k3 << k4),7and the overall process can be treated as the reversible first-order reaction:
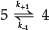
where k+1 and k-1 are the overall rate constants for the forward and reverse reactions respectively, and the overall rate coefficient is the sum of the rate constants for the opposing reactions.
The integrated rate equation for such a reversible first-order reaction can be expressed in the form of Equation 1,7 in terms of which a plot of ln ([5]_[5]eq) vs. t affords a straight line of slope _(k+1 + k-1). Equations 2 and 3 can then be used to determine the individual rate constants.
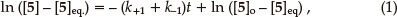
where [5]o is the initial concentration and [ 5]eq the equilibrium concentration.
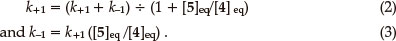
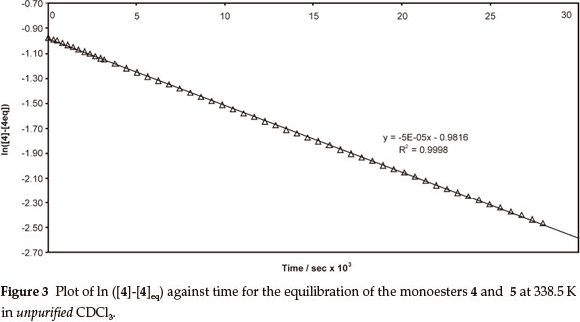
Thus, a plot of the data for the isomerization of compound 5 in unpurified CDCl3 at 338.5 K affords a straight line (e.g. Fig. 3; R2 = 0.9998), from the slope (-4.91 x 10-5) of which the forward and reverse rate constants could be determined [ k+1 = 2.58 (±0.08) x 10-5 s-1 and K-1 = 2.33 (±0.05) x 10-5 s-1, respectively]. When the kinetic study was repeated at 338.5 K using the iso-meric substrate 4, a variation of <1 % from the mean of the k+1 and k-1 values was observed.
Data for the isomerization of the monoester 4 were obtained at eight different temperatures in the range, 302_338.5 K, and an Arrhenius plot (R2 = 0.9422) permitted determination of the activation energy (Ea = 77±7kJmol_1). Similar treatment of the data (R2 = 0.9458) for the reverse reaction (using monoester 5) gave Ea = 78 (± 7) kJmol1. The calculated8values for the enthalpy (ΔΗ‡), entropy (ΔS‡) and Gibbs free energy (ΔG‡) of activation for the forward and reverse reactions at 298 K are summarized in Table 1 (Entries 1 and 2). At all temperatures, the rate of the forward reaction is slightly greater than that of the reverse reaction _ a result consistent with the observed equilibrium concentrations. The large negative entropy changes for the formation of the transition state are, at least, consistent with the formation of a transient cyclic intermediate such as 7 (and/or 7'; Scheme 2).
Acidic impurities are known to develop on exposure of CDCl3 to atmospheric conditions,9 and attention was given to the possible role of acid catalysis in the trans-esterification process (Scheme 3). Consequently, isomerization of the monoesters 4 and 5 was explored under dry N2, at different temperatures, using CDCl3, which had been purified by passage through activated alumina and crushed 4Å molecular sieves to remove practically all traces of HCl and H2O.9 While the rate of the forward reaction was still ca. 10 % faster than the reverse reaction (Table 1; Entries 3 and 4), both processes were of the order of 25 times slower than those in unpurified CDCl3. Moreover, the activation energies (Ea) and the free energies (ΔG‡) and enthalpies of activation (ΔΗ‡) were >100 kJ higher than the corresponding values in unpurified CDCl3. These increases in Ea, ΔΗ‡ and ΔG‡ clearly reflect the absence of significant acid catalysis in the reactions conducted in purified CDCl3.Onthe other hand, the entropies of activation (ΔS‡) for both sets of reactions are indistinguishable within the limits of error, suggesting that, even in the absence of the acid catalyst, the activated complexes possess a similar degree of order. Attempts to quantify the concentration of acid in CDCl3 proved frustrat-ingly difficult, while addition of a small volume of HCl to purified CDCl3 resulted in rapid isomerization and concomitant conjugate addition of HCl to the acrylate moiety. When the experiments were conducted using purified CDCl3 in air, the reactions proceeded, initially, at rates comparable to those observed using purified CDCl3under N2. However, the rates then gradually increased becoming comparable to those for reactions in unpurified CDCl3, the general trend being the higher the reaction temperature, the sooner the increase in the rate _ an observation consistent with the degradation of the CDCl3 in air in these cases. However, the linearity of the pseudo-first-order kinetic plots (e.g. Fig. 3; R2 = 0.9998) is consistent with negligible variation of the acid concentration during kinetic runs using unpurified CDCl3. These observations have led us to conclude that trans- esterification in these systems is catalyzed by trace, but seemingly stable concentrations of acid in the stored (unpurified) CDCl3.
The use of purified10 DMSO-d6 as solvent was expected to obviate the acidic complications observed with CDCl3, and trans-esterification under N2 in this medium was exceedingly slow (<1 % change in the integral ratios over 12 h at 325.2 K and <5 % over 11 h at 348.5K), but the addition of p-toluenesulfonic acid catalyzed the transformation. Not surprisingly, introduction of a catalytic quantity of conc. H2SO4 accelerated the trans-esterification in purified DMSO-d6but resulted in concomitant degradation of the sample.
It is apparent that the reaction rates are strongly influenced by the medium and increase in the order: purified DMSO-d6 < purified CDCl3 < purified DMSO-d6 with p-TsOH < unpurified CDCl3. The activation energies (Ea) for both monoesters, in a given medium, are very similar, as are the activation parameters, ΔΗ‡, ΔG‡ and ΔS‡. Irrespective of the medium, however, the entropies of activation (ΔS‡) are all negative and comparable in magnitude. We thus conclude that the observed trans-esterification proceeds via concurrent, acid-catalyzed, pseudo-first-order, forward and reverse reactions, involving ordered transition-state species. These conclusions are accommodated by the mechanistic proposals outlined in Scheme 3.
Experimental
NMR spectra were recorded on a Bruker AVANCE 400 MHz spectrometer at 305 K. Low-resolution (EI) mass spectra were obtained on a Finnigan-Mat GCQ mass spectrometer and high-resolution (EI) mass spectra on a VG70-SEQ double-focusing magnetic sector spectrometer (Department of Chemistry, University of the Witwatersrand). Optically pure compounds were derived from commercially available, homochiral, (1R)-(+)-camphor. Compounds 211 and 312 are known.
2-exo-3-exo-Bornanyl diacrylate 6,2-exo-hydroxy-3-exo-bornanyl acrylate 4 and 3-exo-hydroxy-2-exo-bornanyl acrylate 5: Butyllithium (1.6 M solution in hexane; 9.62 mL, 15.4 mmol) was added dropwise over 20 min, under dry N2 to a stirred solution of bornane-2,3-diol 3 (2.02 g, 11.8 mmol) in dry THF (25 mL) at -5 °C. After 1.5 h, acryloyl chloride (1.41 g, 15.6 mmol) was added dropwise, the mixture was stirred at 0 °C for 1 h and then allowed to warm to room temperature over 14 h. The THF was removed in vacuo and the residue was treated with satd. aq. NaHCO3 (10 mL), extracted with Et2O (3 x 10 mL) and dried over anhydrous MgSO4. Concentration under reduced pressure gave an oil, which was chromatographed [radial chromatography, using silica gel, 4 mm plate; elution with hexane-EtOAc (9:1)] to afford the following three products.
2-exo-3-exo-Bornanyl diacrylate 6, as white crystals (0.36 g, 11 %) (Found: M+, 278.15199. C16H22O4 requires M, 278.15181); δh (400 MHz; CDCy 0.85,0.88 and 1.15 (9H,3xs, 8-, 9- and 10-Me), 1.19_1.83 (4H, complex of multiplets, 5- and 6-CH2), 1.91 (1H, d, J 4.9 Hz, 4-H), 4.91 (2H, s, 2-H and 3-H), 5.77 and 6.03 (4H, 2 x m, 3'- and 3"-¾) and 6.30 (2H, m, 2' and 2"-H); δc (100 MHz; CDCl3) 10.9, 20.4 and 20.9 (C-8, C-9 and C-10), 23.8 (C-6), 32.9 (C-5), 47.5 and 48.8 (C-1 and C-7), 49.6 (C-4), 77.1 (C-2), 79.8 (C-3), 128.4 (C-2'), 128.5 (C-2"), 130.47(C-3'), 130.54 (C-3"), 164.89 (C-1') and 165.00 (C-1"); m/z 278 (M+, 0.4 %) and 206 (100).
2-exo-Hydroxy-3-exo-bornanyl acrylate 4, as an oil (0.90 g, 34 %) (Found: M+, 224.14161. C13H20O3 requires M, 224.14124); δh (400 MHz; CDQ3) 0.82 (3H, s, 9-Me), 0.93 (3H, s, 8-Me), 1.02_1.84 (4H, complex of multiplets, 5- and 6-CH2), 1.12 (3H, s, 10-Me), 1.84 (1H, d, J 5.0 Hz, 2-OH), 1.88 (1H, d, J 4.9 Hz, 4-H), 3.81 (1H, dd, J 5.2 and 6.2 Hz, 2-Hendo), 4.68 (1H, d, J 6.8 Hz, 3-Hendo), 5.83 (1H, dd, J 1.4 and 10.4 Hz, 3'-Hz), 6.13 (1H, dd, J 10.4 and 17.3 Hz, 2'-H) and 6.39 (1H, dd, J 1.4 and 17.3 Hz, 3-He); δc (100 MHz; CDCl3) 11.0 (C-8), 20.7 (C-10), 21.1 (C-9), 23.9 (C-5), 33.2 (C-6), 46.9 (C-7), 49.0 (C-1), 49.2 (C-4), 79.6 (C-2), 80.0 (C-3), 128.4 (C-2'), 130.7 (C-3') and 166.2 (C-1').
3-exo-Hydroxy-2-exo-bornanyl acrylate 5, as an oil (1.27 g, 48 %) (Found: M+, 224.14161. C13H20O3 requires M, 224.14124); δh (400 MHz; CDCl3) 0.82 (3H, s, 9-Me), 0.90 (3H, s, 8-Me), 1.01_1.75 (4H, complex of multiplets, 5- and 6-CH2), 1.15 (3H, s, 10-Me), 1.79 (1H, d, J 4.8 Hz, 4-H), 1.87 (1H, d, J 4.1 Hz, 3-OH), 4.02 (1H, dd, J 3.7 and 6.2 Hz, 3-Hendo), 4.54 (1H, d, J 6.7 Hz, 2-Hendo), 5.84 (1H, dd, J 0.9 and 10.4 Hz, 3'-Hz), 6.15 (1H, dd, J 10.4 and 17.3 Hz, 2'-H) and 6.40 (1H, dd, J 1.4 and 17.3 Hz, 3-He); δc (100 MHz; CDC3) 11.2 (C-8), 20.6 (C-10), 21.1 (C-9), 24.1 (C-5), 33.1 (C-6), 47.0 (C-7), 48.3 (C-1), 51.6 (C-4), 76.7 (C-3), 82.9 (C-2), 128.4 (C-3'), 130.7 (C-2') and 166.4 (C-1').
Kinetic studies: the monoesters 4 and 5 were purified by HPLC [elution with hexane-EtOAc (7:3)]. The solvent was removed from each fraction under reduced pressure and the purified compounds were stored under N2 at -78 °C. Aliquots (0.5 mL) of solutions of 4 or 5 in deuterated solvent were used for NMR spectroscopic analysis. Data were acquired using an automated programme to obtain spectra at regular time intervals. Final equilibrium concentrations were determined by monitoring the samples, kept in sealed NMR tubes at constant temperature (±0.5 K) for periods of 7-30 days until the product ratios were constant. The NMR data sets were analyzed using a specially developed program13 to process the F.I.D. data and integrate the selected signals. Corrected NMR probe temperatures were used to calculate the kinetic parameters summarized in Table 1.
Acknowledgements
The authors thank the National Research Foundation (NRF; GUN 2069255) and Rhodes University for generous financial support, and Professor M.E. Brown for helpful advice and Dr Z. Tshentu for assistance with the attempted quantification of acid concentrations.
References and Notes
1 See, for example, D. Basavaiah, A.J. Rao and T Satyanarayana, Chem. Rev. 2003, 103, 811. [ Links ]
2 See O.B. Familoni, P.J. Klaas, VE. Pakade and P.T. Kaye, Org. Biomol. Chem., 2006, 4, 3960, and references cited therein. [ Links ]
3 (a) K-S. Yang and K. Chen, Org. Lett, 2000, 2, 729; [ Links ] (b) L.J. Brzezinski, S. Rafel and J.W. Leahy, Tetrahedron, 1997, 53, 16423. [ Links ]
4 See, for example, P.T. Kaye and WE. Molema, J. Chem. Soc, Chem. Commun. 1998, 2479; [ Links ] and M.D. Evans and P.T. Kaye, Synth. Commun., 1999, 29, 2137. [ Links ]
5 VK. Aggarwal, S.Y. Fulford, and G.C. Lloyd-Jones, Angew. Chem. Int. Edn, 2005, 44, 1706. [ Links ]
6 B.S. Furniss, A.J. Hammond, P.W.G. Smith and A.R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, 5th edn., Longman, Harlow, 1989, 626-630. [ Links ]
7 R.B. Jordan, Reaction Mechanisms of Inorganic and Organometallic Systems, 2nd edn., Oxford University Press, 1998, p. 7. [ Links ]
8 J. March, Advanced Oranic Chemistry: Reactions, Mechanisms and Structure, 4th edn., John Wiley and Sons, New York, 1992, p. 225. [ Links ]
9 R.M. Silverstein and F.X. Webster, Spectrometric Identification of Organic Compounds, 6th edn., John Wiley and Sons, New York, 1998, p. 163. [ Links ]
10 DMSO-d6 was purified by passage through neutral alumina and crushed 4 Å molecular sieves (to remove the H2O invariably present). [ Links ]
11 B. Pfrunder and C.H. Tamma, Helv. Chim. Acta 1969, 52, 1632. [ Links ]
12 Beilstein's Handbuch der organishen Chemie, 6, p. 755. [ Links ]
13 K.A. Lobb, Unpublished computer program, Rhodes University, Grahamstown, 2002. [ Links ]
Received 10 December 2012
Accepted 12 April 2013
* To whom correspondence should be addressed. E-mail: p.kaye@ru.ac.za


{kind=link}