










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban ago. 2013
RESEARCH ARTICLE
Inorganic-Organic Hybrids Incorporating a Chiral Cyclic Ammonium Cation
Andreas Lemmerer; David G. Billing
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg, WITS 2050, South Africa
ABSTRACT
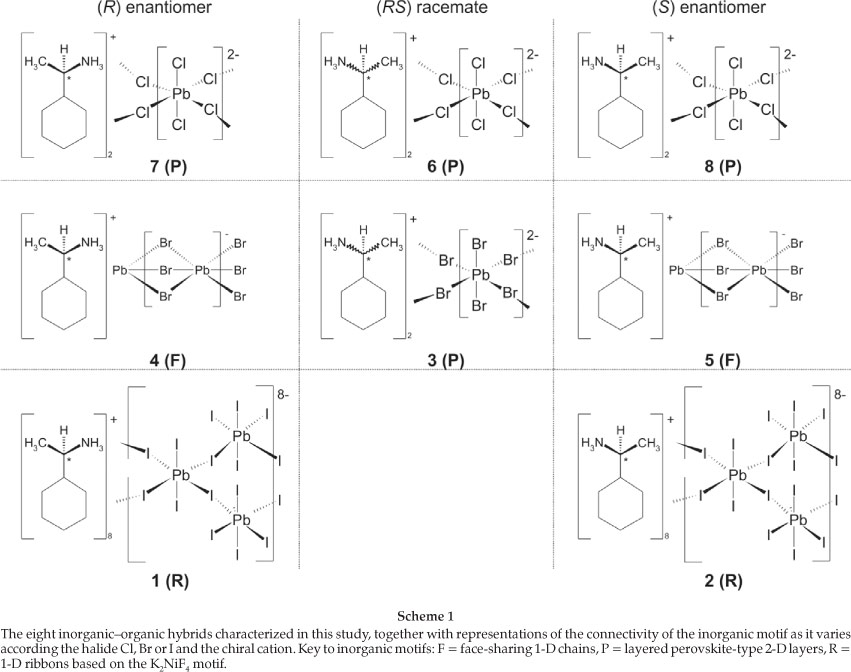
In this paper we report the synthesis and the crystal structure of eight inorganic-organic hybrids containing various lead halides as the inorganic motif and a chiral, primary ammonium cation as the organic constituent. The organic cation investigated is (C6H11C*H(CH3)NH3)+ and both the (R) and (S) as well as the racemic (RS) forms were used. Within the structures obtained, three different inorganic motifs are displayed by the lead halide octahedra: 1-D polymeric face-sharing chains of formula ((R)-[C6H5CH(CH3)NH3)PbBr3](4) and [((S)-C6H5CH(CH3)NH3)PbBr3](5); 1-D polymeric corner-sharing ribbons based on the terminated-K2NiF4 type structure of formula ((R)-[C6H5CH(CH3)NH3)8Pb8I14](1) and [((S)-C6H5CH(CH3)NH3)8Pb8I14] (2); and 2-D corner-sharing layers based on the K2NiF4 perovskite structure type of formula [((RS)-C6H5CH(CH3)NH3)2PbBr4] (3), [((RS)C5H5CH(CH3)NH3)2PbCl4] (6), (R)-[C6H5CH(CH3)NH3)2PbCl4] (7) and [((S)-C6H5CH(CH3)NH3)2PbCl4] (8).
Keywords: Inorganic-organic hybrids, X-ray crystal structure, hydrogen bonding, lead halide octahedra.
1. Introduction
A tenet of crystal engineering is the directed assembly of mole-cules in the crystalline state to a desired structural arrangement that will predispose the compound with certain physical and chemical properties.1 In the set of compounds classified as inor-ganic-organic hybrids, which consist of anionic metal halide octahedra assembled together with an organic ammonium cation, general formula [(R-NH3)xMyXz], where M is any divalent metal and X = Cl, Br and I, a number of different motifs of the inor-ganic component are possible. The choice of the ammonium cat-ion, which can be anything from long alkylammonium chains to complex aromatic structures, influences the motif of the inorganic component. The KjNiF4 type motif, also refered to as the layered perovskite-type motif in the literature where x = 2;y = 1; z = 4, is the most sought after as these compounds form natural quantum well materials that show photoluminescence, electroluminescence and nonlinear optical properties.2 By using the same metal, in this case lead, and only varying the identity of the halide and the R group, we have found a great variety of structures already.3 When the R organic moiety is chiral, the choice of racemic or pure enantiomeric forms in the crystal structure modifies the packing of the layers in the hybrid system. Similiarly, by varying the halide between chloride, bromide and iodide while using the same enantiomer has an effect on which inorganic motif is prefered. This idea was first put into effect recently and three different inorganic motifs were observed.4 The ammonium cation chosen in that study had an aromatic ring system and a chiral functional group as a ring substituent. In this study, we exchange the phenyl for a cyclohexane ring as the R component, and vary the identity of the halide and choose between the optically active enantiomer and the racemate (Scheme 1).
2. Experimental
2.1. Materials
All reagents and solvents employed were commercially avail-able and used as received without further purification. The controlled heating and cooling of the solutions was done with a programmable temperature controller connected to an oil bath (see supplementary information).
2.2. Synthesis
2.2.1. Preparation of [((R)-C6H11CH(CH3)NH3)8Pb3I14], 1 0.059 g PbI2 (0.130 mmol) was dissolved in 2 mL 47 % HI in a large sample vial. Thereafter, 0.025 g (R)-C6H5CH(CH3)NH2 (0.196 mmol) was added and the precipitate dissolved by keep-ing the solution for 12 hours at 80 °C. The solution was slowly cooled to room temperature by 2 °C/h. A yellow single crystal suitable for X-ray diffraction analysis was selected and studied.
2.2.2. Preparation of [((S)-C6H11CH(CH3)NH3)8Pb3I14], 2
0.080 g PbI2 (0.174 mmol) was dissolved in 3 mL 47 % HI in a large sample vial. Thereafter, 0.040 g (S)-C6H5CH(CH3)NH2 (0.314 mmol) was added and the precipitate dissolved by keep-ing the solution for 12 hours at 80 °C. The solution was slowly cooled to room temperature by 2 °C/h. A yellow single crystal suitable for X-ray diffraction analysis was selected and studied.
2.2.3. Preparation of [((RS)-C6H11CH(CH3)NH3)2PbBr4], 3
0.072gPbBr2(0.196mmol) was dissolved in 3mL48% HBr in a large sample vial. Thereafter, 0.014 g (R)-C6H11CH(CH3)NH2 (0.109 mmol) and 0.014 g (S)-C6H11CH(CH3)NH2 (0.109 mmol) was added and the precipitate dissolved by ultrasound. The solution was left at room temperature and crystals grown by slow evaporation. A single colourless crystal suitable for X-ray diffraction analysis was selected and studied.
2.2.4. Preparation of [((R)-C6H11CH(CH3)NH3)1PbBr3], 4
0.090 g PbBr2 (0.245 mmol) was dissolved in 2 mL 48 % HBr in a large sample vial. Thereafter, 0.030 g (R)-C6H11CH(CH3)NH2 (0.234 mmol) was added and the precipitate dissolved by keep-ing the solution for 1 hour at 80 °C. The solution was slowly cooled at 2 °C/h to room temperature. A single colourless crystal suitable for X-ray diffraction analysis was selected and studied.
2.2.5 Preparation of [((S)-C6H11CH(CH3)NH3)1PbBr3], 5
0.081 g PbBr2 (0.221 mmol) was dissolved in 2 mL 48 % HBr in a large sample vial. Thereafter, 0.038 g (S)-C6H11CH(CH3)NH2 (0.296 mmol) was added and the precipitate dissolved by refluxing by keeping the solution for 1 hour at 80 °C. The solution was slowly cooled at 2 °C/h to room temperature. A single colourless crystal suitable for X-ray diffraction analysis was selected and studied.
2.2.6. Preparation of [((RS)-C6H11CH(CH3)NH3)2PbCl4], 6
0.090 g PbCl2 (0.324 mmol) was dissolved in 2 mL 33 % HCl in a large sample vial. Thereafter, 0.030 g (R)-C6H11CH(CH3)NH2 (0.234 mmol) and 0.030 g (S)-C6H11CH(CH3)NH2 (0.234 mmol) was added and the precipitate dissolved by keeping the solution for 1 hour at 80 °C. The solution was slowly cooled at 2 °C/h to room temperature. A single colourless crystal suitable for X-ray diffraction analysis was selected and studied.
2.2.7. Preparation of [((R)-C6H11CH(CH3)NH3)2PbCl4], 7 0.040 gPbCl2(0.144mmol)was dissolved in 3mL33 % HCl in a large sample vial. Thereafter, 0.038 g (R)-C6H5CH(CH3)NH2 (0.296 mmol) was added and the precipitate dissolved by ultra-sound using a Transsonic 460/H ultrasound bath. The solution was left at room temperature and crystals grown by slow evapo-ration. A single colourless crystal suitable for X-ray diffraction analysis was selected and studied.
2.2.7. Preparation of [((S)-C6H11CH(CH3)NH3)2PbCl4], 8 0.050 gPbCl2(0.180 mmol)was dissolved in 4mL 33 % HCl in a large sample vial. Thereafter, 0.047 g (S)-C6H5CH(CH3)NH2 (0.369 mmol) was added and the precipitate dissolved by ultra-sound. The solution was left at room temperature and crystals grown by slow evaporation. A single colourless crystal suit-able for X-ray diffraction analysis was selected and studied.
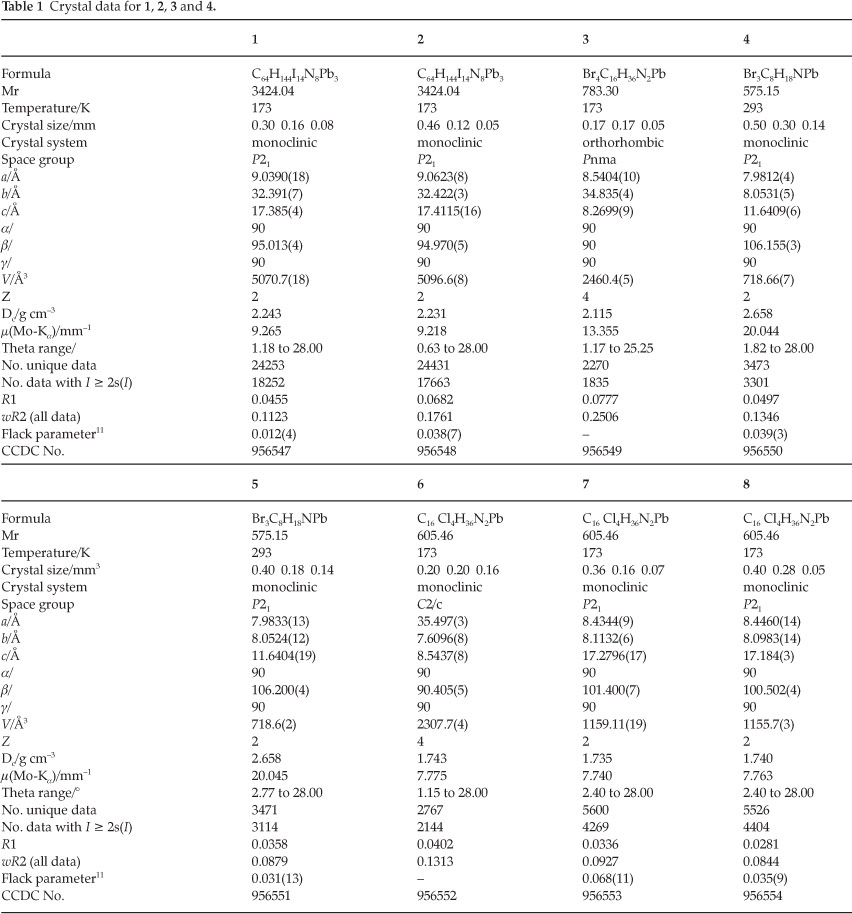
2.3. Crystal Data and X-ray Structure Analysis
Intensity data were collected on a Bruker SMART 1KCCD area detector diffractometer with graphite monochromated Mo Ka radiation (50 kV, 30 mA). The collection method involved w-scans of width 0.3 °. Data reduction was carried out using the program SAINT+, version 6.02.5 and face indexed absorption corrections were made using the program PREP.5
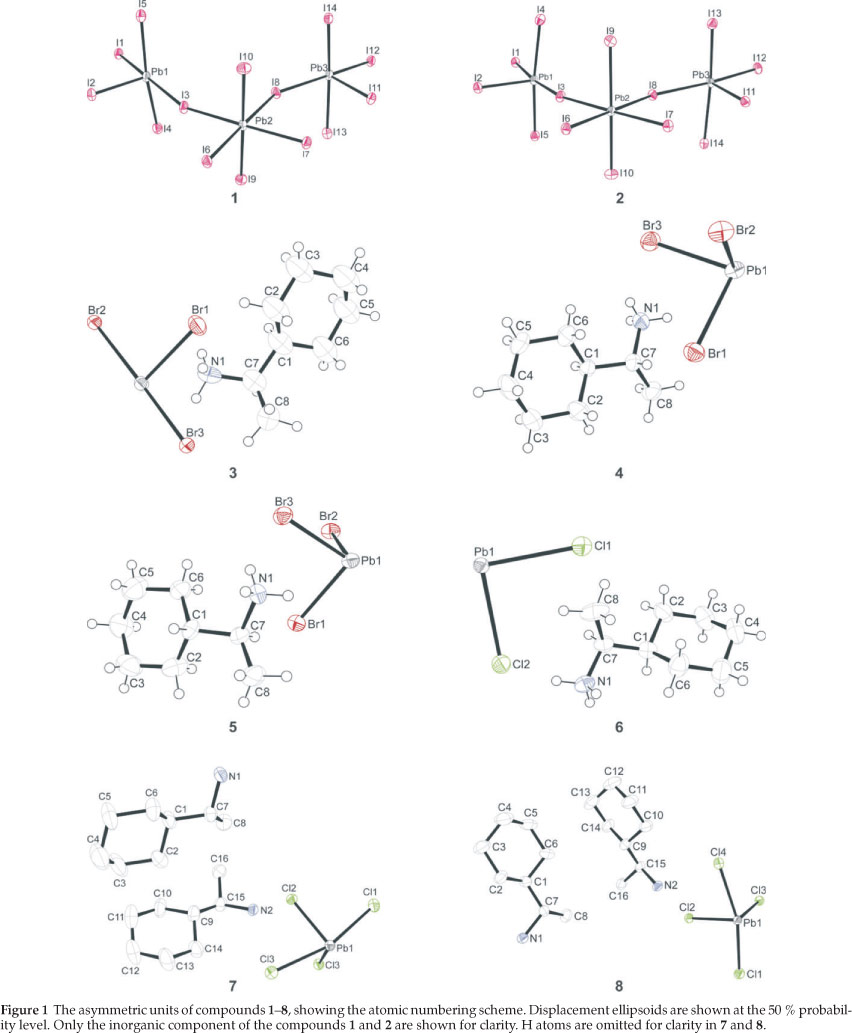
The crystal structure was solved by direct methods using SHELXS-97.6 Non-hydrogen atoms were first refined isotropically followed by anisotropic refinement by full matrix least-squares calculations based on F2 using SHELXL-97.6 Hydrogen atoms were first located in the difference map then positioned geomet-rically and allowed to ride on their respective parent atoms. Diagrams and publication material were generated using WinGX,7 ORTEP8, PLATON9 and DIAMOND.10
The conformational disorder around the cyclohexane ring of cation 8 (see crystal structure description in results for description of cation labelling) in structures 1 and 2 was resolved by finding alternate positions from the difference Fourier map for the respective atoms. Four of the atoms of the six-membered ring are disordered, with the remaining two atoms, C8C and C8F, common to both ring systems. The atoms were then refined anisotropically together with their site occupancy such that the sum of the occupancies for the eight alternate atom positions equaled one. Hydrogen atom positions were then calculated for the respective atoms using a riding model. The ratio of major component, consisting of the atoms C8D, C8E, C8G and C8H, to minor component, consisting of the atoms C8I, C8J, C8K and C8L, is 58.65 to 41.35 % in 1 and 54.73 to 45.27 % in 2.
The 20 0C structures of compounds 1, 2, 3, 6, 7 and 8 were considered to be not good enough for publication. These were subsequently repeated at -100 °C. Comparison of the 20 °C and -100 °C structures of these compounds reveal that no phase changes took place upon cooling. CCDC 956547-956554 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at http://www.ccdc. cam.ac.uk/products/csd/request.html [or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44(0)1223-336033; e-mail: deposit@ccdc.acam.ac.uk. Further crystallographic data are summarized in Table 1 and the atomic numbering scheme and thermal displacement ellipsoids are shown in Fig. 1. All bond distances and angles can be found in the crystallographic information files given as supplementary information.
3. Results and Discussion
Hybrids that have the 2-D layered perovskite-type motif, with general formula [(R-NH3)2MX4], are based on the K2NiF4 or RbAlF4 structure type and result from replacing the K+ and Rb2+ cations with the bulkier ammonium cations. The organic cation can form bilayers between these organic layers if it has only one ammonium group or a monolayer if the organic cation has two ammonium groups. The inorganic layer consists of MX42- corner-sharing metal halide octahedra. Successive inorganic layers can either be staggered relative to each other as in the K2NiF4 structure or be eclipsed, where it is then related to the RbAlF4 structure.12 To keep these larger cations effectively in place, there must exist hydrogen bonding between one end of the organic cation and the halide on the metal. This 'head' of the cation is the primary ammonium group, -NH3+, and the rest of the cation -R is the tail. Weak van der Waals forces between the tails hold the complete sandwich inorganic-organic-inorganic together.
The hydrogen bonding involves all three of the primary ammonium hydrogens. In the 2-D case, two hydrogens can bond to the axial (bridging) halides and the third to the terminal halide (bridging halide configuration) or the reverse case with two terminal and one bridging halide (terminal halide configuration). The hydrogen bonded interactions generally have D...A distances slightly greater than the sum of the van der Waals radii of the ionic species used (Pb2+ = 1.19, Cl- = 1.81, Br- = 1.96 and I- = 2.20 A).13
The alternative inorganic motifs commonly seen are 1-D chains of inorganic metal halides with different connectivity's. The adjacent octahedra in the chains can either share out a single halide (corner-sharing), two halides (edge-sharing) or three halides (face-sharing). The degree of sharing can cause distortion of the geometry of the octahedra with respect to bond angles and bond lengths.
3.1. The Crystal Structures of [((R)-C6H11CH(CH3)NH3)8Pb8I14] (1) and [((S)-C6H11CH(CH3)NH3)8Pb8I14] (2): the Ribbon Motif based on the K2NiF4 Structure Type.
The enantiomorphic pair of compounds, abbreviated [(R)8Pb8I14] and [(S)8Pb8I14], were expected to give layered perovskite-type hybrids as the ammonium cation is sterically similiar to the aromatic equivalent, C6H5CH(CH3)NH3 which gave hybrid perovskites for the R, S and racemic species together with lead iodide.4 Instead, the connectivity has been retained but in place of forming layers, the octahedra form infinite polymeric ribbons, which are three lead atoms abreast, i.e. every fourth row of corner-sharing octahedra is missing from the K2NiF4 structure type (Fig. 2). These chains run along the a-axis. Either side of these chains, there is a gap of 8.34 k between the boundary lead atoms and 4.14 Å between the iodides to the next chain in the c-direction. These are characteristic of distances between adja-cent corner-sharing PbI6 octahedra within the 2-D layered perovskite-type motif. It is these two boundary lead atoms that are surprisingly not bridged by an iodide atom which would then have formed the two-dimensional layer motif (Fig. 3). The organic cations occupy positions above and below the gaps formed by three or four corner-sharing octahedra and the space in between the chains. The chains in 1 and 2 are offset to each other and separated by 4.0560(1) k and 4.0517(1) k, respectively, in the direction of the b-axis. This motif has been seen before only once to the best of our knowledge in the compound [H3N(CH2)3NH3)2Pb1,5Br7.H2O].14
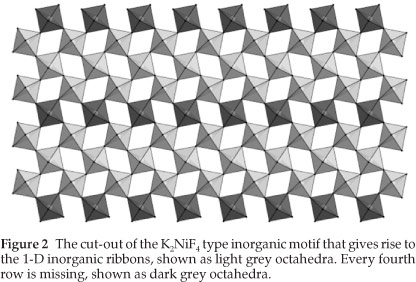
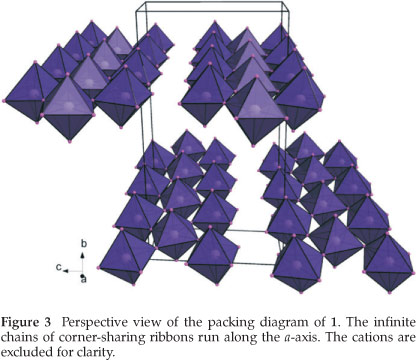
The two structures are enantiomorphs as the inorganic motif is the same regardless of which enantiomer is the counter cation. Compound 1 has three lead atoms in the asymmetric unit which span the breadth of the one-dimensional chain. Each lead atom is bonded to six iodides in a non-ideal octahedral geometry. All six distances to the iodides are different and the cis and trans angles deviate from 90 ° and 180 °. Pb1 and Pb3 occupy the borders of the chain whereas Pb2 is sandwiched in between and runs along the centre of the chains. Hence, Pb2 has four bridging halides as seen in the hybrid perovskite motif while Pb1 and Pb3 have only two bridging halides cis to each other. Pb2 bonds to Pb3 via 17 and 18 and similiarly to Pb1 via 13 and 16. The bridg-ing distances to the outer octahedra are in the range 3.1866(10) A to 3.2122(10) A between Pb2 and 13,16,17 and 18 for structure (1) and 3.1913(15) A to 3.2200(14) A for structure (2). In contrast, the bridging distances for the outer octahedra inwards towards Pb2 are longer. The distance between Pb1 and I3 is 3.3731(10) A (1) and 3.3751(15) A (2) and to I6 3.4425(11) A (1) and 3.4483(15) 2. Pb3 has similiar distances of 3.3862(9) A (1) and 3.3967(15) A (2) and 3.3959(10) A (1) and 3.4047(15) A (2), respectively for I7 and I8. The iodides that are trans to the bridging halides on Pb1 and Pb3, and hence form the boundary of the chain, are shorter, i.e. I12 is trans to I8 and has a distance of 3.0541(10) A (1) and 3.0585(15) A (2)). The same effect is seen in the remaining terminal halides, i.e. I1 (3.1127(10) A (1) and 3.1182(15) (2)), I2 (3.0201(11) A (1) and 3.0305(16) A (2)) and I11 (3.0487(9) A (1), 3.0580(15) A (2)). The remaining six halides, I4 and I5; I9 and I10; I13 and I14, occupy the axial positions of the octahedra with distances in the range 3.2096(10) A to 3.3280(12) A for 1 and 3.1377(18) A to 3.4483(15) A for 2.
An important geometric parameter of layered perovskite-type hybrids is the angle formed by the bridging halide and the two lead atoms it connects. The lead atoms do not occupy positions associated with inversion centres requiring all four bridging angles around Pb2 to be different. There is a difference between the angles that bridge to either side of the chain. The greatest angle is 156.46(3) ° to Pb1 with a slightly shorter angle of 153.46(3) ° via the other halide to the same lead atom. The corre-sponding angles to Pb3 are shorter and differ by 3.65 °.
No less than eight (R)-C6H11CH(CH3)NH3 ammonium cations are in the asymmetric unit, and these are connected to the inor-ganic layers by charge assisted N-H...I hydrogen bonds. The organic cations are labelled cat1 (containing atom N1), cat2 (N2), etc. The cyclohexane rings exist in the chair configuration for all eight, whereas the torsion angles between the rings and the chiral functional groups (given by C3D-C3C-C3A-N3 for cat3 and similarly for all other cations by the letters A-H) are all different which explains the lack of higher symmetry elements between the eight moieties. One ammonium cation is disordered over two positions at right angles to another. The two rings both share the common atoms C8C and C8F.
The interplanar angle between a mean plane formed by the layer of lead and equatorial iodides and a meanplane through the six carbons of the cylohexanes vary between seven of the eight amines. There is a correlation between the torsion angle and the angle between the mean planes. If the torsion angles are close to 180 °, the cations are tilted at a greater angle away from the inorganic layer. Cation 3, 4 and 6 have torsion angles of -177.6(10) °,174.0(1) ° and -169.0(13) ° and respective tilt at angles of 76.01(38) °, 62.31(42) ° and 79.03(46) ° to the layers. The remaining four cations (1, 2, 5 and 7) have torsion angles in the range 52.6(17) ° to 89.5(15) ° and subsequently their tilt angles are close to perpendicular (79.5(5) ° to 87.7(4) °).
The hydrogen bonding can be classified in two categories: if the number of hydrogen bonds to equatorial halides dominate, it is called equatorial halogen configuration and if the number of hydrogen bonds to axial halides dominate, it is called axial halogen configuration.
Cat1 and cat8 occupy the central gaps formed by four joined corner-sharing PbI6 octahedra. This void is characteristic of the hybrid perovskite motif. Both NH3+ groups favour the axial halogen configuration. Cat3 and cat6 sit in the channels between two chains and sit closer to the plane of the chains as there is less steric hindrance. The equatorial configuration is adopted. The remaining four cations (2, 4, 5, 7) occupy the outer voids formed by three PbI6 octahedra only as they sit on the edge of the chains and both apical (2, 5) and equatorial (4, 7) configurations are observed. The hydrogen bonding details for 1 and 2 are given as supplementary information (Table S1 and S2, respectively) and illustrated in Fig. 4 for 1. Compound 2 has similar hydrogen bonding geometries.
3.2. The Crystal Structure of [((RS)-C6HnCH(CH3)NH3)2PbBr4] (3): the K2NiF4 Motif with Staggered Inorganic Layers
Figure 5a clearly underlines a bidimensional arrangement in which a double layer of (R)-1-cyclohexylethylammonium and (S)-1-cyclohexylethylammonium cations are embedded between two consecutive inorganic [PbBr6] sheets, forming an alternated inorganic-organic layered structure. The lead atoms are not aligned from layer to layer, resulting in a staggered arrangement of adjacent layers. In the direction perpendicular to the layers, the primary interaction between the layers are N-H...Br hydrogen bonds. In the direction parallel to the layers, the cohesion is achieved by strong ionic bonds between equatorial bromide and lead atoms.
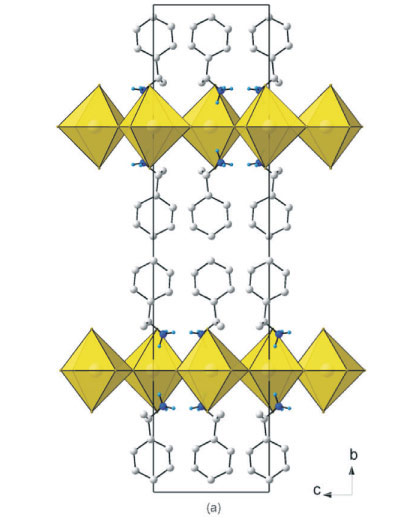
The inorganic layer is built up from characteristic corner-sharing PbBr6 octahedra. The asymmetric unit consists of a lead atom on a special position and three bromide atoms, Br1 occupy-ing the axial position and Br2 and Br3 occupying the equatorial position in the octahedra, with the same special position as the lead atom. The individual PbBr6 octahedra are rotated by 149.92(11) ° and 155.49(11) ° relative to each other. Furthermore, the perovskite-type layers are corrugated in the c-direction by an angle of 6.39(5) ° with respect to the ac-plane. The coordination geometry around the Pb atom shows axial compression of the octahedral geometry, with the average bridging Pb1-Br2 and Pb1-Br3 distances longer than the axial distances Pb1-Br1. The angle between cis and trans related bromide atoms deviate from 90 ° and 180 °, respectively. The racemic 1-cyclohexylethyl-ammonium cation is on a general position. The cyclohexane rings are ordered within the layers and adopt the chair configuration. The cyclohexane rings of adjacent cations are almost parallel (0.4(2) °). The torsion angle H7-C7-C1-H1 is -53.2(3) ° so that the chiral functional group is bent out of the plane of the rings. The angle between adjacent cations is 44.0(1) ° (plane defined by all eight atoms). The hydrogen bonds between the organic and inorganic entities adopt the terminal halogen configuration (see Table 3). The hydrogen acceptor distances to the terminal halide Br1 are 2.48 k and 2.52 k and to the bridging halide Br3 2.69 k.
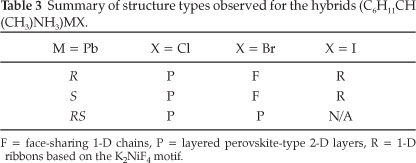
3.3. The Crystal Structures of [((R)-C6HnCH(CH3)NH3)PbBr3] (4) and [((S)-C6H11CH(CH3)NH3)PbBr3] (5): the Face-sharing Motif
These two compounds, abbreviated [(R)PbBr3](4) and [(S)PbBr3](5), are enantiomorphs and possess the same inorganic motif and a similiar packing arrangement of their organic cations. Figure 6a clearly underlines the one-dimensional arrangement of 4, in which four chains of face-sharing PbBr6 octahedra run along the four corners of the unit cell, parallel to the b-axis. In total, there is one complete chain in each unit cell. The channels in between the chains are occupied by (R)-C6HUCH(CH3)NH3 cations in 4 and (S)-C6HUCH(CH3)NH3 in 5. Adjacent chains are stabilised by N-H...Br hydrogen bonds. In the direction parallel to the layers, the cohesion is achieved by strong ionic bonds between the bromide and lead atoms.
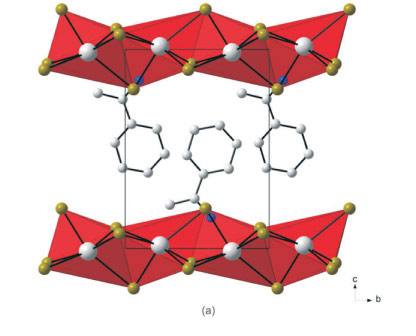
The inorganic motif is built up from characteristic face-sharing PbBr6 octahedra, which form extended chains along the b-axis. The asymmetric unit consists of a lead atom and three bromide atoms, Br1 occupying the axial position with Br2 and Br3 occupying the equatorial positions in the octahedra (Fig. 6b). Within the chains, the shared face consists of these three halides. The octahedra are severely distorted with all lead bromide distances different, ranging from 2.8455(14) Å to 3.3251(14) Å in 4, and from 2.8502(11) Å to 3.3236(12) Å in 5. The bond angles between cis ligands vary from 77.78(4) ° to 116.22(4) ° in 4 and from 78.25(3) ° to 116.15(3) ° in 5. Since the lead atom lies on a general position unlike in structure 3, the three trans angles do not equal 180 ° but range from 160.67(4) ° to 169.22(4) ° in 4 and from 160.71(3) ° to 169.22(3) ° in 5.
The (R)-1-cyclohexylethylammonium cation is on a general position. The unit cell contains two cations and the dihedral angle formed by planes through the rings is 3.02(9) ° in 4 and 3.27(9) ° in 5. The cyclohexane ring adopts the chair conforma-tion. The chiral functional group is aligned with the ring. This is evidenced by a torsion angle of -179.4(1) ° and 178.7(8) ° for H7-C7-C1-H1, respectively for 4 and 5. The plane formed by the cyclohexane rings is in line with the chains of lead atoms. This was measured by measuring the angle between the vector connecting the lead atoms and a least-squares plane through the ring atoms. The angles are 89.3(5) ° for 4 and 88.6(4) ° for 5. In the similar study with the phenyl as the R group, the same inorganic motif was observed for the lead bromides but the R groups are angled differently to the chains. The phenyl rings are angled at 43.0(1) ° for [((R)-1-phenethylammonium)PbBr3j3 and 42.6(2) ° for [((S)-1-phenethylammonium)PbBr3]1b to the face-sharing chains. A possible reason is the possibility of weak π-π interactions between the p systems of adjacent rings, which is absent in the saturated hydrocarbon rings used in this study. The hydrogen bonds between the organic and inorganic entities adopt the equatorial configuration (see Fig. 6b). Two hydrogens bond to the equatorial halides Br2 and Br3 and the third hydrogen bonds to the axial halide Br1. Details of the hydrogen bonding are given in Table 2 for 4 and 5. Due to the geometry of the NH3 group, the hydrogen bonds to the equatorial halides are to one chain and to the axial halide on an adjacent chain.
3.4. The Crystal Structure of [((RS)-C6HnCH(CH3)NH3)2PbCl4] (6): the K2NiF4 Motif with Staggered Inorganic Layers
The crystal structure of 6 has the same layered perovskite-type motif to 3 as the unit cell contains two complete inorganic layers, i.e. adjacent inorganic layers that are staggered relative to each other.
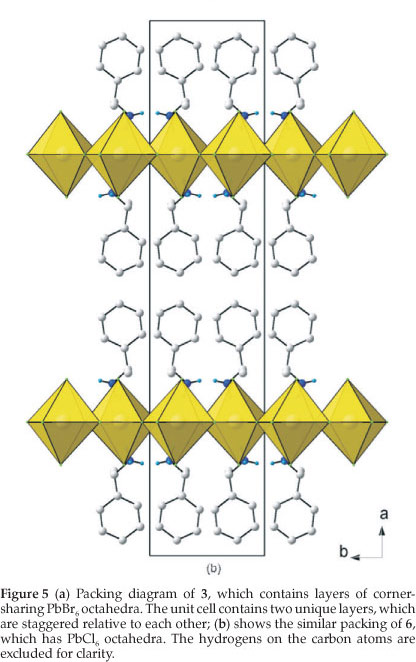
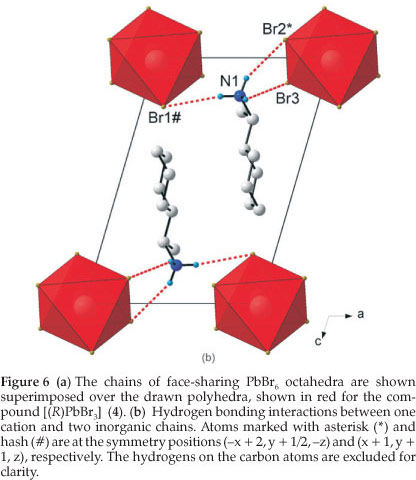
Figure 5b clearly underlines the bidimensional arrangement in which a double layer of (R)-1-cyclohexylethylammonium and (S)-1-cyclohexylethylammonium cations are embedded between two consecutive inorganic [PbCl6] sheets, forming an alternated inorganic-organic layered structure. The lead atoms are not aligned from layer to layer, resulting in a staggered arrangement of adjacent layers.
The inorganic layer is built up from characteristic corner-sharing PbCl6 octahedra. The asymmetric unit consists of a lead atom on a special position and two chloride atoms, Cl1 occupying the axial position and Cl2 occupying the equatorial position in the octahedra. Both chlorides are on general positions. There is one less halide in the asymmetric unit than in 3. This means that there is only one unique bridging halide. The PbCl6 octahedra are rotated by 153.40(8) ° relative to each other. Furthermore, the perovskite-type layers are corrugated in the bc-direction by an angle of 5.26(1) ° with respect to the bc-plane. The coordination geometry around the Pb atom shows axial compression of the octahedral geometry, with the bridging Pb1-Cl2 distances longer (2.920(2) Å and 2.958(2) Å) than the axial distances Pb1-Cl1 (2.845(2) Å). The angle between cis related chloride atoms range from 83.555(19) ° to 96.445(19) °, with all trans angles equal to 180 °, unlike the range of trans angles seen in 3.
The 1-cyclohexylethylammonium cation is on a general position. The cyclohexane rings are ordered within the layers and adopt the chair configuration. Adjacent cations are rotated at an angle of 12.3(4) ° to each other; however the cyclohexane rings themselves are almost parallel to each other as measured by planes through the cyclohexane rings between adjacent molecules (0.1(4) °) as the torsion angle H7-C7-C1-H1 is -61(1)) °. The N-H...Cl hydrogen bonds between the organic and inorganic entities adopt the terminal halogen configuration (see Table 2).
3.5. The Crystal Structures of [((R)-C6HnCH(CH3)NH3)2PbCl4] (7) and [((S)-C6H11CH(CH3)NH3)2PbCl4] (8): the RbAlF4 Motif with Eclipsed Inorganic Layers
The two compounds with pure enantiomers crystallized in the layered perovskite-type motif based on the RbAlF4 structure type in an almost identical fashion. The two structures, abbrevi-ated [(R)2PbCl4](7) and [(S)2PbCl4](8), are enantiomorphic and only differ in the stereochemistry of the ammonium cation. The detailed crystal structure of 7 will be given and deviations and similiarities between the two structures commented on.
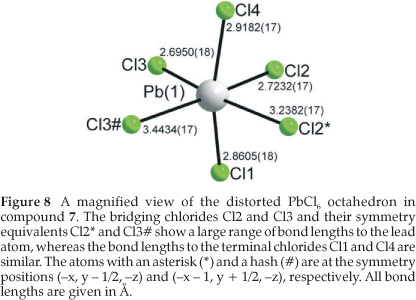
Figure 7a clearly underlines a bidimensional arrangement in which a double layer of (R)-1-cyclohexylethylammonium cations are embedded between two consecutive inorganic [PbCl6] sheets, forming an alternated inorganic-organic layered structure. The lead atoms are aligned from layer to layer, result-ing in an eclipsed arrangement of adjacent layers, typical of monoclinic unit cells. Here lies the main difference between the hybrid perovskite motif adopted in 7 and 8 to the structures 3 and 6, which crystallized in orthorhombic unit cells. Instead of a single unique organic moiety, the use of the optically active enantiomer instead of the racemate as in 6, results in two organic cations observed in the asymmetric unit, as well as one complete PbCl42- unit. The inorganic layer is built up from characteristic corner-sharing PbCl6 octahedra. The asymmetric unit consists of a lead atom Pb1 and four chloride atoms on general positions, Cl1 and Cl4 occupying the axial positions and Cl2 and Cl3 occu-pying the equatorial position in the octahedra. As a result of the lead atom not occupying a special position, the lead atoms do not sit on a plane but deviate by 0.25(2) Å from the ab-plane in 7 and by 0.22(3) Å in 8. As shown in the projection perpendicular to the layers, along the a-axis in Fig. 7b, the PbCl6 octahedra are rotated by 146.12(7) ° and 155.57(7) ° relative to each other when bridged by Cl2 and Cl3, respectively (145.99(5) ° and 155.40(6) ° in 8). The layers are corrugated by 8.18(3) ° (7) and 8.01(3) ° (8) along the a-axis, about 3 ° more than in the racemic layered hybrids 3 and 6. The coordination geometry around the Pb atom shows only a slight axial compression of the octahedral geometry, with the average bridging distances (3.0245(17) Å) longer than the average axial distances (2.8894(17) Å). However, the octahedra are severely distorted in the equatorial plane as there are two long and two short Pb-Cl bond lengths between trans related bridging chlorides (Fig. 8). The difference in the bond lengths are 0.5432(17) Å and 0.7272(18) Å in the same direction. The same phenomena is seen in 8, where the differences are 0.5150(14) k and 0.7708(14) Å. Such a severe disparity is rarely seen in other lead chloride perovskite-type hybrids. A search on the Cambridge Structural database (Version 5.32, November 201015) evidenced five other compounds that have the layered perovskite-type motif with lead chloride: [(C2H5NH3)2PbCl4],16 [(C3H7NH3)2PbCl4]17 and [(CH5C2H4NH3)2PbCl4],18 [(C6H11NH3)2PbCl4]3L and [(o-H3NC6H4NH3)PbCl4].19 The maximum ranges of bond lengths in the lead chloride octahedra are 0.645 A, 0.064, 0.280, 0.283 and 0.610 A, respectively, for those five compounds. It is surprising that the smallest cation of the three, C2H5NH3+, would have the most distorted octahedral geometry. The bond angles between cis and trans related chlorides within the octahedra all deviate from ideality for both 7 and 8. The cis angles in the former structure vary from 81.25(5) ° to 98.85(5) °. The trans angles between the bridging chlorides Cl2 and Cl3 deviate less from 180 ° (172.52(5) ° and 177.65(5) °) than between the axial chlorides Cl1 and Cl4 (163.606 °). In the latter case, the cis angles in 8 vary from 81.15(4) ° to 99.10(4) °. The trans angles between the bridging chlorides Cl2 and Cl3 deviate less from 180 ° (172.67(4) ° and 177.51(4) °) than between the axial chlorides Cl1 and Cl4 (163.33(5) °).
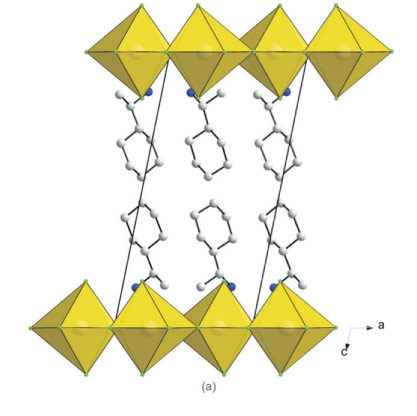
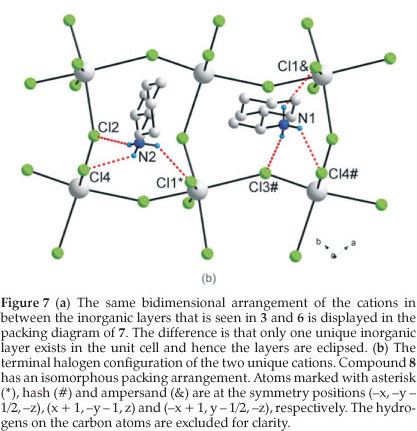
The two (R)-l-cyclohexylethylammonium cations in 7 sit on general positions. The cations are labelled cat1 (containing atom N1) and cat2 (N2). The two cations in the asymmetric unit are not quite perpendicular to each other (82.2(2) °) to coincide with the two orientations of the voids. The cyclohexane rings are ordered within the layers and adopt the chair configuration. The chiral functional groups are parallel to the cyclohexane rings. The torsion angles are -178.6(7)° (catl: H7-C7-C1-H1) and 178.6(7) ° (cat2: H15-C15-C9-H9). The two (S)-1-cyclohexylethyl-ammonium cations in 8 are on general positions and have simi-lar conformational features as to the (R)-1-cyclohexylethyl-ammonium cation in 7.
The hydrogen bonds between the organic and inorganic entities adopt the terminal configuration for both chiral cations, typical of hybrid perovskites with a monoclinic unit cell (see Table 2). The hydrogen acceptor distances to the terminal halides are longer (2.50 Å and 2.52 Å) and (2.57 Å and 2.57 Å) than to the bridging halides (2.42 Å and 2.54 Å), respectively for cat1 and cat2 (Fig. 7b).
The hydrogen bonding between the organic and inorganic entities adopt the terminal halogen configuration for both chiral cations, typical of layered perovskite-type hybrids with a mono-clinic unit cell (See Table 2). The hydrogen acceptor distances to the terminal halides are longer (2.51 Å and 2.54 Á) and (2.56 Å and 2.58 Å) than to the bridging halides (2.41 Á and 2.56 Å) respectively for cat1 and cat2.
4. Conclusion
The objective of this study was to observe the effect on the inorganic motif in the structure of the inorganic-organic hybrids [(C6H11CH(CH3)NH3)xPbyXz] by varying the identity of the halide between chlorine, bromine and iodine and simultaneously, the identity of the chiral organic component.
A different pattern of the type of inorganic motifs that are templated by the choice of halide and chirality of the organic cation was found in this study, summarized in Table 3. In the previous study,3 the layered perovskite-type motif, based on the
K2NiF4 structure type, was adopted when using lead(II) iodide and the (R) and (S) cation and the RbAlF4 structure type in the racemic cation. Here, the compounds 6,7 and 8, which consist of lead(II) chloride octahedra, have the desired 2-D motif. However, the racemic cation produced eclipsed inorganic layers in lead iodide and staggered layers in lead chloride, with the reverse scenario observed for the pure enantiomers: staggered in lead iodide and eclipsed in lead chloride.
The motif adopted by lead iodide in this study is still related to the K2NiF4 motif. Here corner-sharing, 1-D ribbons form chains along one of the axis for both R and S enantiomers, respectively compounds 1 and 2. Attempts at growing crystals of the racemate and lead iodide gave orange crystals of poor quality. The authors surmise that the colour is indicative of the layered perovskite motif, as observed in previous studies of lead iodide hybrids.4
The only common motif observed in both studies was 1-D chains of face-sharing octahedra, observed in both studies for lead bromide and the pure enantiomers, (R)(4) and (S)(5). Lead bromide with the racemic cation, compound 3, has an isostructural packing arrangement and motif to the lead chloride compound, both having staggered inorganic layers. In contrast, the previous study had isostructural 1-D corner-sharing chains for both lead chloride and lead bromide. This study, in conjunction with the previous one, highlight again the unpredictability of and diversity within the field of inorganic-organic hybrids.
References
1 G.R. Desiraju, J. Chem. Sci,. 2010,122, 667-675. [ Links ]
2 (a) D.B. Mitzi, Prog. Inorg. Chem., 1999, 48, 1-121; [ Links ] (b) G.C. Papavassiliou, G.A. Mousdis and I.B. Koutselas, Adv. Mater. Opt. Electron., 1999, 8, 265-271; [ Links ] (c) S.A. Bourne and Z. Mangombo, CrystEngComm, 2004, 6, 437-442.
3 (a) D.G. Billing, Acta Cryst., 2002, e58, m669-m671; [ Links ] (b) D.G. Billing and A. Lemmerer, Acta Cryst., 2003, e59, m381-m383; [ Links ] (c) D.G. Billing and A. Lemmerer, Acta Cryst., 2004, c60, m224-m226; [ Links ] (d) A. Lemmerer and D.G. Billing, Acta Cryst., 2006, e62, m779-m781; [ Links ] (e) A. Lemmerer and D.G. Billing, Acta Cryst., 2006, e62, m904-m906; [ Links ] (f) D.G. Billing and A. Lemmerer, Acta Cryst., 2006, c62, m174-m176; [ Links ] (g) D.G. Billing and A Lemmerer, Acta Cryst., 2006, e62, m1103-m1105; [ Links ] (h) D.G. Billing and A. Lemmerer, Acta Cryst., 2004, c62, m238-m240; [ Links ] (i) D.G. Billing and A. Lemmerer, Acta Cryst., 2004, c62, m264-m266; [ Links ] (j) D.G. Billing and A. Lemmerer, Acta Cryst., 2004, c62, m269-m271; [ Links ] (k) D.G. Billing and A. Lemmerer, CrystEngComm, 2007, 9, 236-244; [ Links ] (l) D.G. Billing and A. Lemmerer, CrystEngComm, 2009, 11, 1549-1562; [ Links ] (m) A. Lemmerer and D.G. Billing, CrystEng-Comm, 2010, 12, 1290-1301; [ Links ] (n) A. Lemmerer and D.G. Billing, CrystEngComm, 2012,14, 1954-1966.
4 D.G. Billing and A. Lemmerer, CrystEngComm, 2006, 8, 686-695. [ Links ]
5 Bruker, SAINT+. Version 6.02 (includes PREP and SADABS). Bruker AXS Inc., Madison, Wisconsin, U.S.A. [ Links ]
6 G.M. Sheldrick, Acta Cryst., 2007, a64, 112-122. [ Links ]
7 L.J. Farrugia, J. Appl. Cryst., 1997, 30, 565. [ Links ]
8 L.J. Farrugia, J. Appl. Cryst., 1999, 32, 837-838. [ Links ]
9 A.L. Spek, J. Appl. Cryst., 2003, 36, 7-13. [ Links ]
10 K. Brandenburg, Diamond. Version 2.1e., Crystal Impact GbR, Bonn, Germany. [ Links ]
11 H.D. Flack, Acta Cryst., 1983, A39, 876-881. [ Links ]
12 (a) D.M. Hatch, H.T. Stokes, K.S. Aleksandrov and S.V. Miguel, Phys. Rev. B, 1989,39,9282-9288; [ Links ] (b) D.M. Hatch and H.T. Stokes, Phys. Rev. B, 1987, 35, 8509-8516. [ Links ]
13 R.D. Shannon, Acta Crystallogr. Sect. A: Fundam. Crystallogr., 1976, 32, 751-767. [ Links ]
14 A.B. Corradi, A.M. Ferrari, G.C. Pellacani, A. Saccani, F. Sandrolini and P. Sgarabotto, Inorg. Chem., 1999, 38, 716-721. [ Links ]
15 F.H. Allen, Acta Cryst., 2002, b58, 380-388. [ Links ]
16 M. Geselle and H. Fuess, Z. Kristallogr. - New Cryst. Struct. (1997), 212, 241. [ Links ]
17 A. Meresse and A. Daoud, Acta Cryst., 1989, c45, 194-196. [ Links ]
18 D.B. Mitzi, J. Solid State Chem., 1999, 145, 694-704. [ Links ]
19 L. Dobrzycki and K. Wo niak, CrystEngComm, 2008,10, 577-589. [ Links ]
 Correspondence:
Correspondence:
E-mail: dave.billing@wits.ac.za
Received 14 November 2011
Revised 3 January 2013
Accepted 7 August 2013


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}