










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban ago. 2013
RESEARCH ARTICLE
Synthesis and Antimicrobial Activities of Some New Pyrazoles, Oxadiazoles and Isoxazole Bearing Benzofuran Moiety
Naqui-Jahan SiddiquiI; Mohammad IdreesI; N.T. KhatiII; M. G. DhondeIII
IDepartment of Chemistry, Government Science College, Gadchiroli, Maharashtra, India
IIDepartment of Applied Chemistry, Priyadarshini Engineering College, Nagpur, India
IIIDepartment of Chemistry, Shri Mathuradas Mohota College of Science, Nagpur, (M.S.), India
ABSTRACT
The synthesis of novel derivatives of pyrazole-3-carboxylate (3-5) from methyl 4-(benzofuran-2-yl)-2,4-dioxobutanoate (1)is reported. Synthesis of substituted 1,3,4-oxadiazoles (7-11) and 5-amino pyrazole-4-carboxylate (12) derivatives starting from the 5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazide (6) are also described. Twelve new compounds were synthesized and their identities have been established on the basis of elemental and spectroscopic analysis such as IR, 1H NMR, 13C NMR, Mass Spectra. The compounds were also screened for their antibacterial and antifungal activities against Gram-positive, Gram-negative bacteria and a fungus.
Keywords: 2,4-Dioxobutanoate, isoxazole, pyrazoles, pyrazole-3-carbohydrazide, 1,3,4-oxadiazoles.
1. Introduction
2,4-Dioxobutanoate derivatives have been found to be very reactive towards organic reagents such as hydroxyl amine hydrochloride, semicarbazide hydrochloride, hydrazine hydrate and phenyl hydrazine, hence they are utilized for the synthesis of substituted isoxazoles and pyrazole carboxylates. Compounds having a pyrazole nucleus are known to possess some important pharmacological activities such as antitumor,1-4 antibacterial,5 fungicidal,6,7 antidiuretic,8 anticancer,9 potent antidiabetic agent,10 anti-inflammatory,11 antidepressant,12,13 and antiviral14 activities. Some substituted pyrazoles are cycloxy-genes-2-(Cox2) selective inhibitors.15 A literature survey indi-cated that pyrazole carboxylates when reacted with hydrazine hydrate yield pyrazole carbohydrazides16,17 possessing interest-ing bioactivities such as antifungal18,19, antimalarial,20 anti-convulsant,21 antituberculosis22,23 and anticancer.24
Pyrazole carbohydrazide reacts with different reagents to give 1,3,4-oxadiazoles which have a broad spectrum of biological and industrial activities.25,26 Among the biological applications reported for 1,3,4-oxadiazoles are hypnotic,27 anticancer,28 anti tuberculostatic,29 antimalarial,30 antimicrobial,31,32 antiviral,33,34 hypoglycaemic,35 anti-HIV activity,36 insecticidal,37 and anti-fungal38 activities. In view of these reports and in continuation of our previous work39 we describe here a facile synthesis of isoxazole and pyrazole-3-carboxylates from methyl 4-(benzo-furan-2-yl)-2,4-dioxobutanoate 1. Simultaneously, we have extended the reactions of 5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazide 6 with different reagents to afford new 1,3,4-oxadiazoles and 5-amino pyrazole-4-carboxylate derivatives by adapting previously reported procedures.17
2. Results and Discussion
The syntheses of the title compounds 1-12 are described in Schemes 1 and 2. At every stage the reaction was monitored with TLC. The identities of these synthesized compounds have been established on the basis of elemental analysis and spectral data such as IR, 1H NMR, 13C NMR and Mass Spectra and they were also screened for their antimicrobial activities. The synthe-sis of the starting compound, methyl 4-(benzo-furan-2-yl)-2,4-dioxobutanoate 1 achieved in quantitative yields by the reference method.39 The IR spectrum of 1 showed the enolic -OH stretch at 3475 cm-1 and C=O stretching in ester group at 1759 cm-1. The 1H NMR spectrum showed a singlet39 at δ 14.24 ppm due to -OH proton, singlet at ö 3.95 ppm due to OCH3, multiplet at ö 7.27-7.72 ppm due to aromatic protons and singlet at ö 7.10 ppm confirms vinylic =CH proton. A chemical shift value in 13C NMR is observed at ö 53.3 ppm due to methoxy carbon; the carbon atoms connected to methoxy group are observed at the range of δ 156-167 ppm, signal at δ 167.9 ppm is due to C-1 carbon in C=O of the ester group whereas C-4 carbon in C=O group under the influence of strong electronegative environment appears downfield at δ 181.0 ppm; the aromatic carbons were observed in expected region. The mass spectrum of this product reveals a molecular ion at m/z 247 [M+H] + is in consistent with the molecular formula C13H10O5.
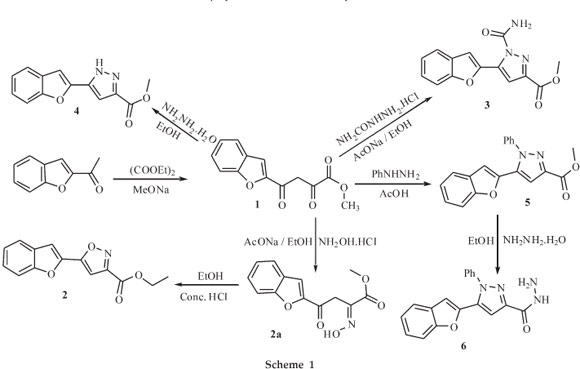
Methyl 4-(benzofuran-2-yl)-2,4-dioxobutanoate 1 on reaction with hydroxyl amine hydrochloride and sodium acetate in absolute ethanol gave methyl 4-(benzofuran-2-yl)-2-(hydroxy-imino)-4-oxobutanoate 2a. The 1H NMR spectrum showed a singlet at δ 12.39 ppm corresponding to enolic -OH proton, multiplet a δ 7.20-7.73 due to aromatic protons, this confirmed that cyclization has not occurred to form an isoxazole ring. Hence, 2a was heated in 50 mL absolute ethanol in presence of conc. HCl for 2 hours to get methyl 5(benzofuran-2-yl)-isoxa-zole-3-carboxylate 2, where a multiplet at δ 7.23-7.75 ppm due to aromatic protons, and a quartet at ö 4.41-4.46, a triplet at δ 1.39-1.42 ppm due to the presence of the COOCH2CH3 group confirmed that trans esterification has also occurred simultaneously.
Treatment of 1 with semicarbazide hydrochloride and sodium acetate in absolute ethanol gave 3, with hydrazine hydrate in acetic acid afforded 4, while its reaction with phenyl hydrazine in acetic acid furnished 5.
Formulation of the reaction products designated 2, 3, 4 and 5 in Scheme 1, was based upon the comparative reactivity of two carbonyl groups in 1. The C-2 carbonyl group being more reactive than the C-4 carbonyl group, the first gets preferably attacked by the nucleophilic reagent such as hydroxylamine hydrochloride, semicarbazide hydrochloride, hydrazine hydrate and phenyl hydrazine to give the corresponding intermediate which simultaneously undergo ring closure with elimination of a water molecule from the imino proton and the -OH group of the enolized C-4carbonyl group forming 2,3,4 and 5. The reaction of 5 with hydrazine hydrate in ethanol gave 5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazide 6. The character-istic IR bands in compounds 3, 4, 5 and 6 at 1694,1693 1618 and 1649 cm-1, respectively shows strong stretching bands due to the C=N group in pyrazole ring.
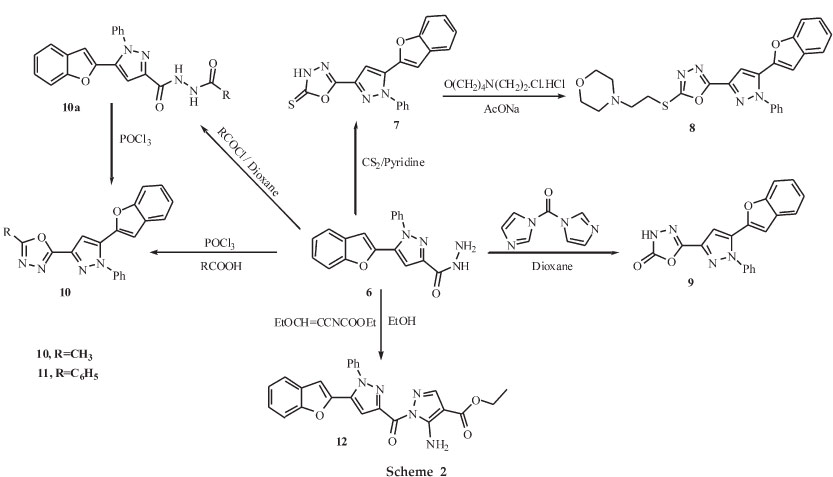
5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazide 6 was further utilized for the synthesis of several innovative azoles 7-12. The interaction of 6 with carbon disulphide in pyridine gave 7. The IR spectra of 7 showed a characteristic absorption band at 3405 cm-1 due to NH; similarly, 1H NMR revealed an exchangeable imino proton at δ 14.79 ppm due to -NH in oxadiazole ring and absence of SH signal indicates its existence as the thione tautomer. The thione function of 7 was alkylated using the bioactive alkylating agent 4-(2-chloroethyl) morpholine hydrochloride in absolute ethanol (99.9 %) in presence of fused sodium acetate to afford 4-(2-(5-(5-(benzo-furan-2-yl)-1-phenyl-1H-pyrazol-3-yl)-1,3,4-oxadiazol-2-ylthio) ethyl) morpholine 8. The 1H NMR spectrum of 8 showed a singlet at δ 6.28 ppm due to CH in pyrazole and a multiplet at δ 7.20-7.55 ppm due to aromatic protons; similarly, a singlet at δ 2.58 ppm due to -CH2NCH2, a singlet at δ 2.88 ppm due to -SCH2CH2N, a singlet at δ 3.53 ppm due to -SCH2CH2N, and a singlet at δ 3.74 ppm due to -CH2OCH2.Inthe 13C NMR spec trum the C-2 and C-5 carbon of the oxadiazole gave signals at 164.40 and 159.95 ppm. The mass spectrum of compound 8 displayed a molecular ion peak at m/z 474 [M+H]+ which is in agreement with the molecular formula C25H23O3N5S.
Treatment of the 6 with N,N'-carbonyldiimidazole (CDI) in dioxane gave 9. The IR spectra of 9 showed a characteristic absorption band at 1768 cm-1 due to C=O. The 1HNMR spectrum showed a singlet at δ 12.38 ppm due to NH of oxadiazole. The reaction of 6 with acetyl chloride in dioxane did not afford the expected product 10. However, the N-acetyl carboxylic acid hydrazide was shown to be the reaction product. The formation of N'-acetyl-5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazide 10a as an intermediate was confirmed from the 1H NMR spectra. A singlet at δ 1.98 ppm due to CH3, one singlet at δ 10.11 ppm and another singlet at δ 10.55 ppm due to the NH protons, a multiplet at δ 7.22-8.08 ppm due to aromatic protons, revealed that cyclization did not occur to give 10. Therefore, 10a was subjected to cyclodehydration in phosphorous oxy-chloride to give the corresponding oxadiazole 10, which was also obtained by heating 6 with acetic acid in POCl3. Similarly, another oxadiazole derivative 11 was obtained by the reaction of 6 with benzoic acid in POCl3. The IR absorption bands in the range of 1695-1600 cm-1 is due to C=N group and bands observed in the range of 1275-1200 and 1075-1020 cm-1 are due to C-O-C grouping of 1,3,4-oxadiazole nucleus of the com-pounds 7-11.
Finally, the reaction of 6 with ethyl(ethoxymethylene) cyano-acetate in ethanol gave the corresponding ethyl 5-amino-1-(5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbonyl)-1H-pyrazole-4-carboxylate 12. The IR spectra of 12 showed a characteristic absorption band at 1697 cm-1 due to C=O stretch. The 1H NMR spectrum showed a triplet at δ 1.32-1.35 ppm for -CH2CH3and quartet at δ 4.25-4.30 ppm for -CH2CH3 singlet at δ 6.3 ppm due to CH in pyrazole and multiplet at δ 7.23-7.63 ppm for thirteen protons including 11 aromatic and two NH2 protons. Its mass spectrum gave a molecular ion at m/z 442[M+H]+ and elemental analysis showed that it is consistent with the molecu-lar formula C24H19O4N5.
Carbon atoms in all the novel synthesized compounds 1-12 from the 13C NMR spectra were seen at their expected chemical shifts; similarly, their mass spectrum revealed a molecular ion peak at m/z [M+H]+ which are in agreement with the molecular formulae of all these synthesized compounds. It was found that the spectral data such as IR, 1H NMR, 13C NMR, and Mass spec-trum of all these newly synthesized compounds in Schemes 1 and 2 were in accordance with the proposed structures.
MIC values are in the range of 15.5-500 mL-1 for both Gram-positive and Gram-negative bacteria was observed (see Table 1). The title compounds were graded as highly active with MIC values of 15.5-62 mL-1, moderately active at 125 mL-1 and poorly active at values 250-500 ug mL-1. The antibacterial screening results revealed that most of the synthesized com-pounds 1-12, exhibited significant antibacterial activities. The test compounds 1, 7, 8 and 12 were found to be highly active, while 2, 5, 10 and 11 were moderately active against B. subtilis. Compounds 4, 5, 8, 10 and 11 were highly active, while 1 and 3 were moderately active against S. aureus. 4, 6, 8 and 10 were highly active but 2, 7 and 12 were moderately active against E. coli. Compound 5 has shown excellent antibacterial activity as compared to the standard drug against E. coli. Compounds 2,10, 11 and 12 were highly active while 1,4,6 and 9 were moderately active against P. aeruginosa. However, all these compounds exhibited significant activity in the range of 31 250 ug mL-1 against A. niger, compounds 1,2,3,6,10 and 12 were found to be highly active while 5, 8 and 11 are moderately active. The rests of the compounds gave poor activities against all the test bacteria and a fungus.
3. Experimental
Melting points were recorded in open capillary in silicon oil bath and are uncorrected. IR spectra were recorded on a Shimadzu IR Spectrophotometer in KBr pellets. 1H NMR and 13C NMR spectra are recorded on a Bruker AM 400 instrument (400 MHz) using tetramethylsilane (TMS) as an internal refer-ence and DMSO-d6/CDCl3 as solvent. Chemical shifts are given in parts per million (ppm). Positive-ion electrospray ionisation (ESI) mass spectra were obtained with a Waters Micromass Q-TOF Micro, Mass Spectrophotometer. Elemental analysis was done using Vario EL III Elemental Analyzer, all compounds showed satisfactory elemental analysis. The reaction was moni-tored by E. Merck TLC aluminum sheet silica gel60F254 and visual-izing the spot in UV light and iodine chamber.
Synthesis of Methyl 4-(Benzofuran2-yl)-2,4-dioxobutanoate (1 )
Diethyloxalate (1.46 mL, 10 mmol) was gradually added with stirring to a solution of 2-acetyl benzofuran (1.6 g, 10 mmol) and sodium methoxide (0.23 g Na in 5 mL methanol, 10 mmol) in N,N-Dimethylformamide (100 mL). The reaction mixture was stirred for 12 h at room temperature, the product obtained was acidified by 1:1 ice-cold HCl, filtered, washed with water and recrystallized from acetone to get yellow crystalline solid 1 (85 %); m.p.: 131-133 °C; R = 0.66 (ethylacetate/n-hexane 1:2); IR (KBr, nmax): 3475 (-OH), 3059, 3020 (ArH), 2968, 2879 (CH3) 1805, 1759 (C=O, ester), 1624,1573,1521 (C=c) cm-1; 1HNMR (CDCl3): δ 3.95 (s, 3H, CH3), 7.10 (s, 1H, =CH), 7.27-7.72 (m, 5H, ArH), 14.24 (s, 1H, -OH) ppm; 13C NMR (CDCl3): 53.3,99.3,112.4,114.1, 123.3, 124.2, 127.3, 128.8 , 150.8, 156.2, 162.3, 167.9, 181.0 ppm; ESI(+)-MS: m/z 247 (M+H)+. Anal. calcd. for C13H10O5: C, 63.41; H, 4.06 %. Found: C, 62.52; H, 4.13 %.
Synthesis of Methyl 5-(Benzofuran-2-yl)-isoxazole-3-carboxylate (2)
Hydroxylamine hydrochloride (1.39 g, 20 mmol) and sodium acetate (1.64 g, 20 mmol) were added to a mixture of methyl 4-(benzofuran-2-yl)-2,4-dioxobutanoate 1 (2.46 g, 10 mmol) in absolute ethanol (99.9 %, 200 mL), and the reaction mixture was refluxed for 4 h. It was concentrated, cooled, poured in ice-cold water and kept overnight; the solid separated out was filtered and recrystallized from diluted ethanol to get 2a, as an interme-diate. Further, 2a was refluxed for2hin absolute ethanol (50 mL) and conc. HCl (1 mL). The solvent was evaporated under reduced pressure to get pale yellow crystalline solid 2 (90 %); recrystallized from ethanol; m.p.: 80-82 °C; Rf = 0.62 (ethylacetate/n-hexane 1:2); IR (KBr, nmax): 3084 (ArH), 2991,2943 (CH3), 1723 (C=O, ester), 1620 (C=N), 1546, 1477, 1459, 1435 (C=C) cm-1; 1H NMR (DMSO-d6): δ 1.39-1.42 (t, J = 8Hz, 3H,-OCH2CH3), 4.41-.46 (q, J = 8 Hz, 2H, -OCH2 CH3), 7.23-7.75 (m, 6H,ArH) ppm; 13C NMR (DMSO-d6): δ 13.8,61.8,101.5,107.1, 111.4, 122.1, 123.7, 126.5, 127.3, 142.7, 154.7, 156.4, 158.7, 162.4 ppm; ESI(+)-MS: m/z (%) 258 (M+H)+; Anal. calcd. for C14H11O4N: C, 65.36; H, 4.28; N, 5.45 %. Found: C, 65.03; H, 4.01; N, 5.39 %.
Synthesis of Methyl 5-(Benzofuran-2-yl)-1-carbamoyl-1H-pyrazole-3-carboxylate (3)
Semicarbazide hydrochloride (1.12 g, 10 mmol) and sodium acetate (0.82 g, 10 mmol) were added to methyl 4-(benzo-furan-2-yl)-2,4-dioxobutanoate 1 (1.23 g, 5 mmol) in absolute ethanol (99.9 %, 10 mL), and the reaction mixture was refluxed for 4 h. It was then concentrated, cooled and poured in ice-cold water, solid separated out was filtered and recrystallized from ethanol to get white crystalline solid 3 (76 %); m.p.: 142-144 °C; Rf = 0.61 (ethylacetate/n-hexane 1:2); IR (KBr, νmax): 3461, 3405, 3255, 3165 (NH2), 3052, 3011 (ArH), 2956, 2919, 2853 (CH3), 1769, 1744 (C=O, ester), 1694 (C=N), 1589 (C=N), 1395 (C-N, amide), 1500, 1436, 1408 (c=C) cm-1; 1H NMR (DMSO-d6): δ 3.95 (s, 3H,-COOCH3), 7.23-7.91 (m, 8H, Ph + NH2) ppm; 13C NMR (DMSO-d6): δ 51.7, 109.1, 111.4, 121.8, 123.1, 125.5, 128.1, 136.1, 143.4, 144.4, 150.2,154.0, 154.1, 161.0 ppm; ESI(+)-MS: m/z (%) 286 (M+H)+; Anal. calcd. for C14H11O4N3: C, 58.94; H, 3.85; N, 14.73 %. Found: C, 58.88; H, 3.68; N, 14.54 %.
Synthesis of Methyl 5-(Benzofuran-2-yl)-1H-pyrazole-3-carboxylate (4)
Hydrazine hydrate (1.5 mL, 30 mmol) was added gradually with constant stirring to methyl 4-(benzofuran-2-yl)-2,4-dioxo-butanoate 1 (2.46 g, 10 mmol) in CH3COOH (30 mL), and refluxed for 2 h. After that it was poured in ice-cold water, filtered and recrystallized from ethanol to get white crystalline solid 4 (90 %); m.p.: 180-182°C; Rf = 0.65 (ethylacetate/n-hexane1:2); IR (KBr, νmax): 3254, 3165 (NH), 3051, 3011 (ArH), 2957 (CH3), 3876 (C=O), 1693 (C=N), 1492, 1453, 1436, (C=C) cm-1; 1H NMR (DMSO-d6): δ 3.91 (s, 3H,-COOCH3), 7.15-7.62 (m, 7H, ArH + NH) ppm; 13C NMR (DMSO-d6): δ 51.53, 102.39, 105.62, 110.77, 120.91, 122.94, 124.35, 128.08, 132.08, 133.09, 141.46, 153.96, 167.28 ppm; ESI(+)-MS: m/z 243 (M +H)+; Anal. calcd. for C13H10O3N2: C, 64.46; H, 4.13; N,11.57 %. Found: C, 64.21; H, 4.34; N, 11.77 %.
Synthesis of Methyl 5-(Benzofuran-2-yl)-1-phenyl-1Ef-pyrazole-3-carboxylate (5)
Phenyl hydrazine (1.62 mL, 15 mmol) was added to a mixture of methyl 4-(benzofuran-2-yl)-2,4-dioxobutanoate 1 (2.46 g, 10 mmol) in CH3COOH (30 mL), and the reaction mixture was refluxed for 4 h. After that it was concentrated and poured in crushed ice, filtered off and recrystallized from acetic acid as white crystalline solid 5(85 %); m.p: 161-163 °C; Rf = 0.63 (ethylacetate/n-hexane 1:2); IR (KBr, νmax): 3061 (ArH), 2955 (CH3), 1618 (C=N), 1734 (C=O, ester), 1593, 1500, 1436, 1408 (C=C) cm-1; 1H NMR (CDCl3) δ: 3.97 (s, 3H, -COOCH3), 6.22 (s, 1H, pyrazole CH), 7.17-7.54 (m, 10H, ArH) ppm; 13C NMR (CDCl3): δ 52.30, 105.62, 109.41, 111.33, 121.44, 123.39, 125.48, 126.40 (2C), 127.93, 129.37 (2C), 129.72, 136.04, 139.53, 144.22, 145.06, 154.48, 162.45 ppm; ESI(+)-MS: m/z 319 (M+H)+; Anal. calcd.forC19H14O3N2: C,71.69;H,4.40;N,8.81 %.Found: C,71.05; H, 4.42; N, 8.42 %.
Synthesis of 5-(Benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazide (6)
Hydrazine hydrate (100 %, 1.7 mL) was added to a mixture of methyl 5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-car-boxylate 5 (3.18 g, 10 mmol) in absolute ethanol (99.9 %, 100 mL), and refluxed for 8 h. It was then concentrated, filtered and recrystallized from ethanol as white crystalline solid 6(88 %); m.p: 145-146 °C; Rf = 0.61 (ethylacetate/n-hexane 1:2); IR (KBr, νmax): 3429,3317,3225,3159 (-NH.-NH2), 3066 (ArH), 1683 (C=O), 1649 (C=N), 1531, 1597 (C = C) cm-1; 1H NMR (CDCl3): δ: 3.69-3.71 (s, 2H, -CONHNH2), 6.22 (s, 1H, pyrazole CH), 7.17-7.54 (m, 10H, ArH), 8.49 (b,1H,-CONHNH2) ppm; 13C NMR (CDCl3): δ 105.68, 107.83, 111.38, 121.40, 123.37, 125.43, 126.05 (2C), 127.92, 129.43 (2C), 129.56, 136.04, 139.54, 145.15, 145.87, 154.51,162.35 ppm; ESI (+)-MS: m/z 319 (M+H)+; Anal. calcd. for C18H14O2N4,: C, 67.92; H, 4.40; N, 17.61 %. Found: C, 67.50; H, 4.35; N, 16.88 %.
Synthesis of 5-(5-(Benzofuran-2-yl)-1-phenyl-1H-pyrazol-3-yl)-1,3,4-oxadiazole-2(3H)-thione (7)
A mixture of 6 (3.18 g, 10 mmol) in CS2 (30 mL) and pyridine (100 mL) was refluxed on water bath for 6 h. Then it was cooled and excess of solvent was removed under reduced pressure. The residue obtained was triturated with ice-water mixture and neutralized with dilute HCl. The solid obtained was filtered and recrystallized from ethanol to get white crystalline solid 7 (95 %); m.p.: 249-250 °C; R = 0.61 (ethylacetate/n-hexane 1:2); IR (KBr, νmax): 3405 (-NH), 3070,3019 (ArH), 1636 (C=N-N=C), 1594,1518, 1497, 1472, 1431 (C=C), 1255, 1072 (C-O-C), 1230 (C=S) cm-1; 1H NMR (DMSO-d6): d 6.5 (s, 1H pyrazole CH), 7.22-7.62 (m, 10H, ArH), 14.79 (s, 1H, -NH) ppm; 13C NMR (DMSO-d6): δ 105.99,106.33,110.91,121.41,123.24,125.41,125.68 (2C), 127.35, 129.23 (2C), 129.58, 135.77, 136.75, 138.83, 144.02, 153.92, 155.53, 177.34 ppm; ESI(+)-MS: m/z 361 (M+H)+; Anal. calcd. for C19H12 O2N4S: C, 63.33; H, 3.33; N, 15.55; S, 8.88 %. Found: C, 63.42; H, 3.30; N, 15.61; S, 8.94 %.
Synthesis of 4-(2-(5-(5-(Benzofuran-2-yl)-1-phenyl-1H-pyrazol-3-yl)-1,3,4-oxadiazol-2-ylthio)ethyl) Morpholine (8)
A mixture of 7 (3.6 g, 10 mmol), sodium acetate (4.1 g, 50 mmol) and morpholine hydrochloride (1.88 g, 10 mmol) in absolute ethanol (99.9 %, 100 mL) was refluxed for 6 h. Then it was cooled, excess of solvent was evaporated under reduced pressure; the residue obtained was triturated with water. Solid obtained was filtered off, recrystallized from ethanol as white crystalline solid 8(89 %); m.p.: 160-162 °C; R = 0.56 (ethylacetate/n-hexane 1:2); IR (KBr, νmax): 3030 (ArH), 1607 (C=N), 1593, 1555, 1518, 1514 (C=C) cm-1; 1H NMR (DMSO-d6): d 2.58 (s, 4H, -CH2NCH2-), 2.88 (s, 2H, SCH2CH,N), 3.53 (s, 2H, -SCH2CH2N), 3.74 (s, 4H, -CH2OCH2-), 6.28 (s, 1H, pyrazole CH), 7.20-7.55 (m, 10H, ArH) ppm; 13C NMR (DMSO-d6): δ 29.89,52.79 (2C), 56.60, 66.11 (2C), 105.95, 106.55, 110.95, 121.47, 123.29, 125.44, 125.78 (2C), 127.40, 129.28 (2C), 129.58, 135.69, 137.42, 138.92, 144.01, 153.91, 159.95,164.40 ppm; ESI(+)-MS: m/z474 (M+H)+; Anal. calcd. for C25H23O3N5S: C,63.42;H,4.86;N,14.79;S,6.76. % Found: C,63.41; H, 4.42; N, 14.88; S, 6.96 %
Synthesis of 5-(5-(Benzofuran-2-yl)-1-phenyl-1H-pyrazol-3-yl)-1,3,4-oxadiazol-2(3H)-one (9)
A mixture of 6 (3.18 g, 10 mmol) and N,N'-carbonyldiimidazole (2.43 g, 15 mmol) in 1,4 dioxane (100 mL) was refluxed for 8 h. The reaction mixture was cooled; residue obtained was triturated with ice-water mixture. The solid obtained was filtered off, recrystallized from ethanol as white crystalline solid 9 (70 %); m.p.: 250-252 °C; Rf = 0.53 (ethylacetate/n-hexane 1:2); IR (KBr, νmax): 3511, 3219, 3153 (-NH), 3115 (ArH), 1768 (C=O), 1627 (C=N), 1594, 1499, 1440 (C=C); 1H NMR (DMSO-d6): δ 6.32 (s, 1H, pyrazole, CH), 7.20-7.69 (m, 10H, ArH), 12.38 (s, 1H, NH) ppm; 13C NMR (DMSO-d6): δ 105.74,105.94,110.96,121.48, 123.30, 125.42, 125.75 (2C), 127.41, 129.27 (2C), 129.48, 135.43, 138.17,138.98,144.25,149.29,153.90,154.06ppm; ESI(+)-MS: m/z 345[(M+H)+, 100]; Anal. calcd. for C19H12O3N4: C, 66.27; H, 3.48; N, 16.28 %. Found: C, 66.08; H, 3.51; N, 16.04 %
Synthesis of 2-(5-(Benzofuran-2-yl)-1-phenyl-1H-pyrazol-3-yl)-5-phenyl-1,3,4-oxadiazole (10)
Method 1: Step I: Synthesis of N'-acetyl-5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazide (10a): A mixture of 6 (3.18 g, 10 mmol) and acetyl chloride (1.18 mL, 15 mmol) in 1,4 dioxane (100 mL) was refluxed for 4 h. The reaction mixture was concentrated, filtered and recrystallized from ethanol to get white crystalline solid 10a (79 %); m.p.: 215-217 °C; 1H NMR (DMSO-d6): d 1.98 (s, 3H, CH3), 6.38 (s, 1H pyrazole -CH), 7.22-8.08 (m, 10H, ArH), 10.11 (s, 1H, NH), 10.55 (s, 1H, NH) ppm.
Step II: Synthesis of(10): The intermediate 10a obtained was then refluxed in (25 mL) phosphorous oxychloride at 100 °C for 5 h. Then it was poured in ice-cold water and neutralized with 20%NH4OH, filtered and recrystallized from ethanol as white crystalline solid 10 (70 %); m.p.: 208-210 °C.
Method 2: Synthesis of (10): A mixture of 6 (1.27 g, 4 mmol) and acetic acid (0.24 mL, 4 mmol) in POCl3 (40 mL) was refluxed for 2 h. Excess of solvent was evaporated under reduced pressure, poured in water and neutralized with NH4OH. The solid obtained was filtered off, and recrystallized from ethanol as white crystalline solid 10 (80 %); m.p.: 208-210 °C; Rf = 0.59 (ethylacetate/n-hexane 1:2); IR (KBr, νmax): 3059 (ArH), 2974,2927 (CH3), 1612, 1594, 1576, 1510, 1441, 1459 (C = c); 1H NMR (DMSO-d6): δ 2.61 (s, 3H, -CH3), 6.56 (s, 1H, pyrazole -CH), 7.22-7.62 (m, 10H, ArH) ppm; 13C NMR (DMSO-d6): δ 10.53, 105.87, 106.54, 110.93, 121.41, 123.24, 125.38, 125.74 (2C), 127.40, 129.23 (2C), 129.50, 135.63, 137.81, 138.96, 144.23, 153.91, 159.61, 163.31 ppm; ESI(+)-MS: m/z 343 (M+H)+; Anal. calcd. for C20H14 O2N4: C, 70.17; H, 4.09; N, 16.37 %. Found: C, 70.20; H, 4.10; N, 16.4 %
Synthesis of 2-(5-(Benzofuran-2-yl)-1-phenyl-1H-pyrazol-3-yl)-5-phenyl-1,3,4-oxadiazole (11)
A mixture of 6 (3.18 g, 10 mmol) and benzoic acid (1.22 g, 10 mmol) in POCl3 (15 mL) was refluxed on steam bath for 6 h. The reaction mixture was cooled and poured on ice-water; Ph of the solution was maintained to 7 by adding NH3. The solid obtained was filtered off and recrystallized from 1,4 dioxane as white crystalline solid 11 (95 %); m.p.: 250-252 °C; Rf = 0.58 (ethylacetate/n-hexane 1:2); IR (KBr, νmax): 3065 (ArH), 1694 (c=N), 1591, 1549, 1461, 1493, 1445 (C=C), 1259 (C-O-C); 1H NMR (DMSO-d6): d 6.50 (s, 1H, pyrazole CH), 7.22-8.18 (m, 15H, ArH) ppm; ESI(+)-MS: m/z 405 (M+H)+; Anal. calcd. for C25H16O2N4: C, 74.25; H, 3.96; N, 13.86 %. Found: C, 74.18; H, 4.02; N, 14.12 %
Synthesis of Ethyl 5-Amino-1-(5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbonyl)-1H-pyrazole-4-carboxylate (12)
A mixture of 6 (3.18 g, 10 mmol) and ethylethoxymethylene cyanoacetate (1.69 g, 10 mmol) in absolute ethanol (99.9 %, 100 mL) was refluxed for 8 h. After cooling the solvent was removed in vacuo, filtered and recrystallized from ethanol as fluffy white solid 8 (75 %); m.p.: 204-205 °C; Rf = 0.60 (ethyl-acetate/n-hexane 1:2); IR (KBr,νmax): 3459,3316,3178 (NH2), 3062, 3036 (ArH), 2975 (CH3), 1697(C=O), 1626, 1597 (C=O), 1557, 1496,1469,1451 (C=C) cm-1; 1H NMR (DMSO-d6): δ 1.32-1.35 (t, J = 6,3H, CH2CH3), 4.25-.30 (q, J = 6.6,2H, CH2CH3), 6.3 (s, 1H, pyrazole -CH), 7.23-7.63 (m, 13H, ArH and NH2) ppm; ESI(+)-MS: m/z 442 (M+H)+; Anal. calcd. for C24H19O4N5: C,67.44; H,4.45;N, 16.39 %. Found: C,67.40; H,4.51; N, 16.19 %.
4. Antimicrobial Activity
All the novel synthesized compounds 1-12 were screened for their in vitro antibacterial activity against two Gram-positive strains, i.e. Bacillus subtilis (NCIM 2439) and Staphylococcus aureus (NCIM 2079) and two Gram-negative strains, i.e. Escherichia coli (NCIM 2064) and Pseudomonas aeruginosa (NCIM 2053) in addition to a fungus Aspergillus niger (NCIM 501). Antibacterial activ-ity was assessed by serial twofold (broth) dilution technique, using Muller-Hinton broth for bacteria and Sabouraud dextrose agar for fungus in the concentration of 1000 ,µg mL-1. Ampicillin was used as a standard drug for bacteria and Clotrimazole for fungus. Similarly, serial dilution tubes for standard drug with its stock solution 100 ,µg mL-1 were also prepared so that the concen-trations of standard drug in five tubes were 50, 25, 12.5, 6, 3/µgmL-1. Antimicrobial activity of dimethyl sulphoxide against the organisms were investigated, it was nil. Double strength nutrient broth was used as a growth media. The stock solution was serially diluted to give concentration of 500-15 ,µg mL-1 in nutrient broth. The inoculums size was approximately 106 colony forming units (CFC mL-1). The inoculated tubes were incubated for 24 h at 37(±1) °C (bacteria) and for 72 h at 28 °C (fungus). After 24 h and 72 h the inoculated culture tubes were macroscopically examined for turbidity. The culture tube showing turbidity (lower concentration) and culture tube showing no turbidity (higher concentration) gave the minimum inhibitory concentration (MIC) for the compound. The antimicrobial activity of all the screened compounds as well as standard drug Ampicillin and Clotrimazole determined in terms of minimum inhibitory concentration (MIC mL-1) are given in Table 1.
5. Conclusions
Methyl4-(benzofuran-2-yl)-2,4-dioxobutanoate 1 was reacted with various nucleophilic reagents such as hydroxylamine hydrochloride, semicarbazide hydrochloride, hydrazine hydrate and phenyl hydrazine to synthesize novel isoxazole and pyrazole-3-carboxylates derivatives. 5-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-3-carbohydrazide 6 was further utilized for the synthesis of newer 1,3,4-oxadiazole and 5-amino pyrazole-4-carboxylate derivative. The compounds exhibited promising antibacterial activity in vitro against both Gram-positive and Gram-negative strains of bacteria along with a fungus.
Acknowledgements
The authors are thankful to the Head, Department of Applied Chemistry, Priyadarshini College of Engineering, and Nagpur for providing Laboratory facilities and to the Director, SAIF, Punjab University, and Chandigarh for providing 1H-NMR, 13C NMR and Mass Spectra, RSIC, CDRI, Lucknow, India for pro-viding CHN analysis. The authors are also thankful to Dr. Ugale, Head, Department of Pharmacology, College of Pharmacy, Kamptee for antimicrobial screening of the compound.
References
1 A.M. Farag, A.S. Mayhoub, S.E. Barakat and A.H. Bayomi, Bioorg. Med. Chem., 2008,16,4569-578. [ Links ]
2 B. Insuasty, A. Tigreros, F. Orozco, J. Quiroga, R. Abonia, M. Nogueras, A. Sanchez and J. Cobo, Bioorg. Med. Chem., 2010, 18, 4965-4974. [ Links ]
3 A.M. Farag A.S. Mayhoub, S.E. Barakat, A.H. Bayomi, Bioorg. Med. Chem., 2008,16, 881-889. [ Links ]
4 I. M. El-Deeb and S.H. Lee, Bioorg. Med. Chem., 2010,18, 3961-3973. [ Links ]
5 B.S. Holla, P.M. Akberali and M.K. Shivananda, IL Farmaco., 2000,55, 256-263. [ Links ]
6 A. Mustafa, C.A. Hismat and M.M.J. Yannis, J. Prakt. Chem., 1970,312, 1011-1019. [ Links ]
7 M.H. Elnagdi, M.R.H. Elmoghayor, E.A.A. Hafez, and H.H. Alnima, J. Org. Chem., 1975,40,2604-2607. [ Links ]
8 H.G. Garg, J. Med. Chem., 1972,15, 446-447. [ Links ]
9 F. Manna, F. Chementi, R. Fioravanti,and A. Bolasco, Bioorg. Med. Chem. Lett., 2005,15, 4632-4635. [ Links ]
10 J.H. Ahn, H.M. Kim, S.H. Jung, S.K. Kang, K.R. Kim, S.D. Rhea, S.D. Yong, H.G. Cheon and S.S. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 4461-4465. [ Links ]
11 R.V. Ragavan, V. Vijayakumar and N.S. Kumari, Eur. J. Med. Chem., 2010,45, 1173-1180. [ Links ]
12 Y.R. Prasad, A.L. Rao, L. Prasoona, K. Murali and K.P. Ravi, Bioorg. Med. Chem. Lett., 2005,15, 5030-5034. [ Links ]
13 J.C. Jung, E.B. Watkins and M.A. Avery, Heterocycles., 2005,65,77-94. [ Links ]
14 A.E. Rashad, M.I. Hegab, R.E. Abdel-Megeid, J.A. Micky and F.M.E. Abdel-Megeid, Bioorg. Med. Chem., 2008,16, 7102-7106. [ Links ]
15 M. Ezawa, D.S. Garvey, D.R. Janero, S.P. Khamapure, L.G. Letts, A. Martino, R.R. Ranetunge, D.J. Schwalb and D.V Young, Lett. Drug Design. Discov, 2005, 2, 40-43. [ Links ]
16 B.F. Abdel-Wahab, H.A. Abdel-Aziz and E.M. Ahmed, Arch. Pharm. Chem. Life Sci., 2008, 341, 734-739. [ Links ]
17 A.R. Farghaly and H. El-Kashef. ARKIVOK, 2006, (xi), 76-90. [ Links ]
18 P. Vicini, F. Zani, P. Cozzini and I. Doytchinova, Eur. J. Med. Chem., 2002, 37, 553-564. [ Links ]
19 C. Loncle, J.M. Brunel, N. Vidal, M. Dherbomez and Y. Letourneux, Eur. J. Med. Chem., 2004, 39,1067-1071. [ Links ]
20 P. Melnyk, V. Leroux, C. Sergheraert and P. Grellier, Bioorg. Med. Chem. Lett., 2006,16, 31-35. [ Links ]
21 K. Sridhar, S.N. Pandeya, J.P. Stables and R. Atmakuru, Eur. J. Pharm. Sci. 2002,16,129-132. [ Links ]
22 B.K. Kaymakcloglu and S. Rollas, Il Farmaco., 2002, 57,595-599. [ Links ]
23 J. Patole, U. Sandbhor , S. Padhye, D.N. Deobagkar, C.E. Anson and A. Powell, Bioorg. Med. Chem. Lett., 2003,13, 51-55. [ Links ]
24 E.G. Chalina and L. Chakarova, Eur. J. Med. Chem., 1998, 33(12), 975-983. [ Links ]
25 P.-F. Xu, Z.-H. Zhang, X.-P. Hui, Z.-Y. Zhang, R.-L. Zheng, J. Chin. Chem. Soc., 2004, 51, 315-319. [ Links ]
26 G. Sahin, E. Palaska, Lu. M. Ekizog, M. Ozalp, Il Farmaco., 2002, 57, 539-542. [ Links ]
27 C.H. Lee, H.I. Cho and K.J. Lee, Bull. Korean Chem. Soc., 2000, 22, 1153-1155. [ Links ]
28 A. Mohsen, M. Omar, and D.A. Wafa, J. Heterocyclic Chem., 1984, 21, 1415-1418. [ Links ]
29 K. Potts, in Comprehensive Heterocyclic Chemistry, (A.R. Katritzky and C. Rees, eds.), vol. 6, Pergamon Press, Oxford, 1984, p. 427. [ Links ]
30 M. Akhatar, A. Husain, B. Azad and M. Ajaml, Eur. J. Med. Chem., 2009, 44, 2372-2378. [ Links ]
31 A.A. El-Emam, O.A. Al-Deeb, M. Al-Omar and Lehmann, J. Bioorg. Med. Chem., 2004,12,5107-5113. [ Links ]
32 S.G. Kügükgüzel, E.E. Oruc, S. Rollas, F. Sahin and A. Ozbek, Eur. J. Med. Chem., 2002, 37,197-206. [ Links ]
33 S.G. Kügükgüzel, A. Kocatepe, E. De Clercq, F. Sahin and M. Güllüce, Eur. J. Med. Chem. 2006, 41, 353-359. [ Links ]
34 T.M.C. Tan, Y. Chen, K.H. Kong, J. Bai, Y. Li, S.G. Lim, T.H. Ang and Y. Lam, Antiviral. Res., 2006, 71, 7-14. [ Links ]
35 A.O. Maslat, M. Abussaud, H. Tashtoush and M. Al-Talib, Pol. J. Pharmacology., 2002, 54,55-59. [ Links ]
36 J. Taoa, L.H. Cao, C.F. Wang and D.Z. Wang, J. Chin. Chem. Soc., 2006, 53(5), 1193-1197. [ Links ]
37 X. Zheng, Z. Li, Y. Wang, W. Chen, Q. Huang, C. Liu and G. Song, J. Fluorine. Chem., 2003,123, 163-169. [ Links ]
38 C.J. Chen, B.-A. Song, S. Yang, G.-F. Xu, P.S. Bhadury, L.-H. Jin, D.-Y. Hu, Q.-Z. Li, W. Xue, P. Lu and Z. Chen, Bioorg. Med. Chem., 2007,15, 3981-3989. [ Links ]
39 N.J. Siddiqui, M. Idrees, N.T. Khati and M.G. Dhonde, Bull. Chem. Soc. Ethio., 2013, 27, 85-94. [ Links ]
 Correspondence:
Correspondence:
E-mail: naquiphd.2010@gmail.com
Received 4 February 2013
Revised 15 July 2013
Accepted 15 August 2013


{kind=link}
{kind=link}