




![Synthesis and characterization of ethyl 2-methyl-7-oxo-7H-isoxazolo[2,3-α ]pyrimidine-6-carboxylate derivatives](/img/es/next.gif)





Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban ago. 2013
RESEARCH ARTICLE
Synthesis of Triazole-linked 2-Trichloromethylquinazolines and Exploration of Their Efficacy Against P falciparum
Anton R. HamannI; Carmen de KockII; Peter J. SmithII; Willem A.L. van OtterloI; Margaret A.L. BlackieII
IDepartment of Chemistry and Polymer Science, University of Stellenbosch, Stellenbosch, 7600, South Africa
IIDepartment of Pharmacology, University of Cape Town, Groote Schuur Hospital, Observatory, 7925, South Africa
ABSTRACT
Using 2-trichloromethylquinazoline as scaffold, seven novel triazole-linked compounds have been synthesized using CuAAC chemistry. The in vitro biological activity of four of the compounds on the Plasmodium falciparum chloroquine-sensitive strain NF54 was then determined. The compounds which were tested showed moderate activity with 1.45 /iM as the lowest inhibitory concentration.
Keywords: Malaria, 2-trichloromethylquinazoline, copper click chemistry, 1,2,3-triazole
1. Introduction
Each year malaria affects between 300 and 500 million people and responsible for the death of around 2.5 million people.1 The emergence of drug resistance in species of the genus Plasmodium towards the frequently used drug molecules such as chloro-quine necessitates the discovery of new molecules with novel mechanisms of action.2 In the late 1970s it was determined that thioquinazolines (1) (Fig. 1) have shown potent antimalarial effects against chloroquine-resistant variants of the Plasmodium species.3 In addition, other quinazoline-derived compounds containing thiourea moieties (2) have been synthesized and confirmed to have antiplasmodial activity.4 More recently, work by Vanelle and co-workers5 on a substituted quinazoline scaffold, namely the 2-trichloromethylquinazoline (3) (Fig. 1), demonstrated promising antimalarial activity for a set of molecules based on this particular scaffold.
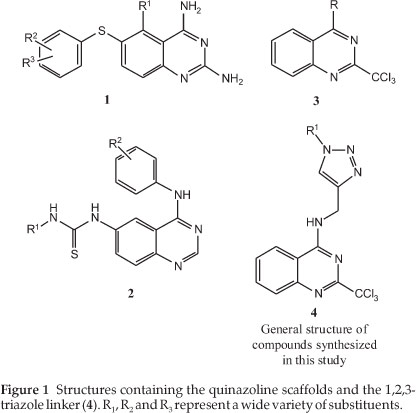
The 1,2,3-triazole moiety is an attractive option to use as a structural feature in medicinal chemistry as it is stable in acidic/ basic as well oxidative/reductive conditions.6 This heterocycle also has a high dipole moment and affords the possibility of forming hydrogen bonds with biomolecular targets.7 These systems are also attracting attention as they are readily synthesized by well-defined copper-catalyzed azide-alkyne cyclization (CuAAC) methods.8,9 Of relevance to this paper is that in recent years, the use of CuAAC and the incorporation of triazole into drug-like molecules with potential antimalarial activity have seen increasing popularity.10-14 In this publication, we have designed and synthesized a small series of novel trichloro-methylquinazolines (4) employing 'click chemistry' as a means to introduce diversity into the side chain. The side chains included unsubstituted aryl groups and some small heterocyclic groups in order to give a starting point for this exploration. The aryl groups were chosen as Vanelle and co-workers5 had used the simpler quinazoline structure 3 with a variety of phenyl groups and achieved micromolar activities against a variety of strains of P. falciparum. The nitrogen-containing side chains were all chosen as a variety of these ligands have been used in quinolines again to good effect.
2. Results and Discussion
2.1. Synthesis
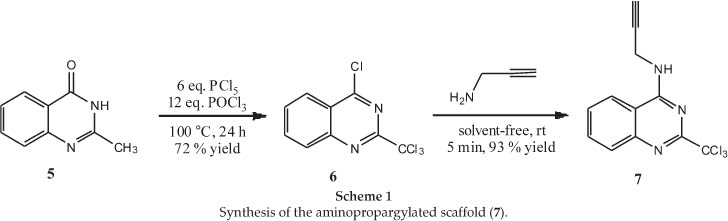
The synthesis of the quinazoline scaffold was based on the procedure described by Vanelle and co-workers using a conventional method,15 followed by a coupling with propargyl amine in a solvent-free medium (Scheme 1)16 to obtain the amino-propargylated scaffold (7) in good yield.
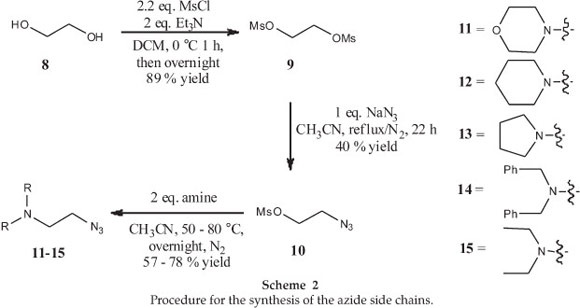
The next step required the synthesis of the substituted azides necessary for the CuAAC reactions. To this end, the synthesis of the azide side chains began with the double mesylation of ethylene glycol (8), followed by the addition of the azide functional group on one side to from compound 10. Five different secondary amines (11-15) were then incorporated by displacing the remaining mesylate group using standard SN2 chemistry (Scheme 2).17 At this point it should be mentioned that the spectra for all the azido compounds generated here compared well with that published in the literature (see experimental section for more details). In addition, the use of IR spectroscopy provided additional evidence that the compounds contained an azide functional group with a characteristic signal being seen at ~2100 cm-1.
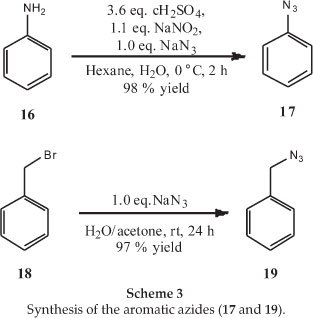
Another two azide side chains with aryl side chains were synthesized to determine whether an aromatic group is necessary for good antimalarial activity, as set out in Scheme 3.18,19 It should be noted that the target of the trichloromethylquinazolines has not yet been established, so it is not clear whether a basic nitrogen in the side chain is necessary, as it is in chloroquinoline-based molecules.20
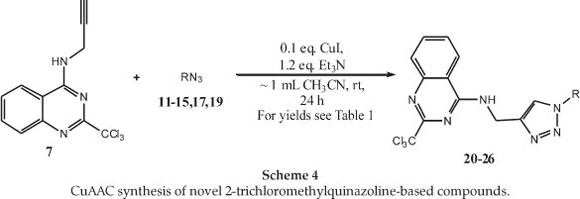
With the alkyne (7) and azide starting materials (11-15,17,19) in hand, the CuAAC reactions, were attempted using the well established conditions.21 CuSO4 was utilized as the source of Cu(II), along with the reducing agent sodium ascorbate, in a protic solvent mixture such as MeOH/H2Oor t-BuOH/H2O. Unfortunately, under these conditions the desired products failed to form, or were only obtained in very low yields, even when using excess copper salt in one of the attempts. In addition, the use of only MeOH or t-BuOH as solvent did not improve the results. As it was noted that Dabiri et al. had obtained acceptable yields using a Cu(OAc)2 salt as the Cu(II) source, this particular copper salt was also tried in our reactions using the above-mentioned solvent systems, but little or no improvements were observed.22
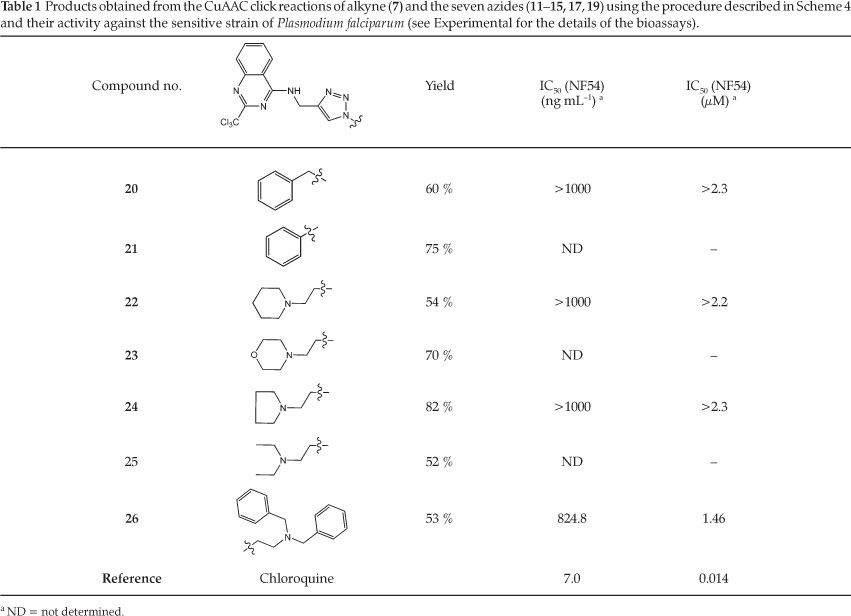
Since the general conditions did not work out as anticipated, an alternative method was tried. The copper catalyst was thus changed to CuI and an aprotic solvent system was used, as adapted per the reaction conditions described by Bernard et al. (Scheme 4).23 In this approach, the yields obtained were significantly higher and the starting material was almost completely consumed. After optimizing the conditions for the click reaction (Scheme 4), the results obtained for the seven azides are tabulated in Table 1. Whilst we have not unequivocally determined the source of the problem in the first reaction conditions, the presence of Cu(II) in a protic solvent appears to favour the formation of a copper complex(es) with (7), thus rendering it less available or inactive for catalysis of the CuAAC reaction. The CuAAC products (21-26) were readily purified by silica gel column chromatography and were obtained in good to moderate yields (52-82 %). The compounds were then fully characterized by NMR and other spectroscopic techniques (see experimental section for details).
The synthesized triazoles were then subjected to in vitro testing using the modified method of Trager and Jensen24 and Makler and co-workers25 on the sensitive P. falciparum NF54 strain. A full dose-response was performed on all compounds to determine the concentration inhibiting 50 % of parasite growth (IC50 value) and the values are shown in Table 1. The results of the in vitro assay against the chloroquine-sensitive strain were mostly in the millimolar range, with compound 26 showing the best activity. Testing out against a chloroquine-resistant strain was not performed as it was thought unlikely that these molecules would show substantially improved efficacy in the resistant strain. This is because the proposed mechanism of action, whilst not unequivocally established, is thought to be different to that of chloroquine. The activities reported by Vanelle and co-workers5 for similar compounds (cf 3 with R = Ph, IC50 against W2 strain was 8.0 µM) can be considered to be within a similar range - low micromolar values. Noting that different strains of the parasite cannot be compared directly. Absolute values of activity for most of these compounds were not determined directly because they were submitted alongside another completely unrelated group of compounds which were substan-tially more active. Expending further effort to determine the efficacy of these compounds was not deemed worthwhile.
In conclusion, a small series of novel compounds has been syn-thesized using a suitably aminopropargylated 2-trichloro-methylquinazoline as a scaffold, and a CuAAC approach. Furthermore, we report a CuAAC method capable of generating the desired triazole-linked entities in the presence of coordinating nitrogen atoms. The problems we had in synthesizing the final compounds were resolved, in part, by using the conditions stated, and a protic solvent, but further work would need to be carried out to investigate the optimum conditions for this reaction. Finally, the compounds were evaluated in terms of their ability to inhibit the P. falciparum NF54 strain parasite, and were found to demonstrate weak activity. It was not clear from these results what modifications of the side chain would be usefully made in order to improve efficacy as there was no marked different between the aryl and heterocyclic side chains. However, it can be noted that the inclusion of the triazole moiety did not appear to have an effect on the efficacy of this class of compounds and therefore can be used to introduce diversity in the side chain. It may be worthwhile to determine if biological activity of 26 can be improved by adding functional groups on the aromatic rings.
3. Experimental
3.1. General
Chemicals were purchased from commercial suppliers and were used as supplied. Melting points were determined in open capillary tubes with a Gallenkamp apparatus and are uncor-rected. 1H and 13C NMR spectra were determined on a Varian Unity Inova 300 MHz or 400 MHz spectrometer at Stellenbosch University. Chemical shifts are given in ö values referenced to the solvent and are reported in parts per million. High-resolution mass spectrometry was performed by CAF (Central Anaytical Facility) at Stellenbosch University using a Waters Synapt G2 spectrometer. Silica Gel 60 was used for flash column chromatography. The progress of the reactions was monitored by thin layer chromatography on using Kieselgel 60 plates.
3.2. Biological Testing
The test samples were tested in triplicate on one or two separate occasions against chloroquine-sensitive (CQS) strain of Plasmodium falciparum (D10). Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method of Trager and Jensen (1976). Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler (1993). The test samples were prepared to a 20 mg mL-1 stock solution in 100 % DMSO and sonicated to enhance solubility. Samples were tested as a suspension if not completely dissolved. Stock solutions were stored at -20 °C. Further dilutions were prepared on the day of the experiment. Chloroquine (CQ) was used as the reference drug in all experiments. A full dose-response was performed for all compounds to determine the concentration inhibiting 50 % of parasite growth (IC50-value). Test samples were tested at a starting concentration of 100 ,µg mL-1, which was then serially diluted twofold in complete medium to give 10 concentrations; with the lowest concentration being 0.2 ,µg mL-1. The same dilution technique was used for all samples. The highest concentration of solvent to which the parasites were exposed to had no measurable effect on the parasite viability (data not shown). The IC50-values were obtained using a non-linear dose-response curve fitting analysis via Graph Pad Prism v.4.0 software.
3.3. Synthesis of Compounds 4-Chloro-2-trichloromethylquinazoline (6)
PCl5 (3.90 g, 18.7 mmol) and POCl3 (9.4 mL, 37 mmol) were added to 2-methylquinazolin-4-one (0.50 g, 3.1 mmol) at 0 °C. The mixture was then heated to reflux and stirred for 24 hours. After cooling, the solution was diluted with CHCl3 (30 mL) and ultra-sound was used to completely dissolve the reaction product. The solution was then added to a beaker of ice and alkalinized with dry NaHCO3 (180 g) while stirring. Alkalinity of the water layer was confirmed with a universal pH indicator.
The solution was filtered to remove the salt and the solution was extracted with CHCl3 (50 mL). The brown organic layer was dried over anhydrous MgSO4, which was removed by filtration and the filtrate concentrated under vacuum. Purification of the brown solid was performed by flash chromatography with CH2Cl2-petroleum ether (1:1) as eluent to give the product as white powder (0.63 g, 72 % yield). Mp 126 °C (neat); IR (ATR): 1558 (m, aromatic), 775 cm-1 (s, CCl3); 1H NMR (CDCl3,300 MHz, 25 °c): ö = 7.87 (ddd, J = 8.3, 7.0 and 1.2 Hz, 1H, CH), 8.09 (ddd, J = 8.5, 7.0 and 1.4 Hz, 1H, CH), 8.27(ddd, J = 8.5,1.2 and 0.7Hz, 1H, CH), 8.34 (ddd, J = 8.4, 1.4 and 0.7 Hz, 1H, CH); 13C NMR (CDCl3, 75.5 MHz, 25 °C): ö = 95.9 (C), 122.9 (C), 125.9 (C), 129.7 (C), 130.6 (C), 135.9 (C), 150.1 (C), 159.9 (C), 164.0 (C). The spectro-scopic data were in agreement with reported data.8
N-(Prop-2-yn-1-yl)-2-(trichloromethyl)quinazolin-4-amine (7)
Propargylamine (0.053 mL, 1.6 mmol) was added dropwise onto 6 (0.15 g, 0.52 mmol). The solvent-free reaction mixture was then stirred at rt for 5 min. The crude residue was then extracted with CH2Cl2. The organic layer was washed with water (3 x 15 mL), dried over anhydrous MgSO4, filtered and concentrated under vacuum. Chromatography with CH2Cl2 as eluent afforded the desired product (7) as white powder (0.14g, 93 % yield). Mp 103 °C (neat); IR (ATR): 3333 (w, NH), 2925 (w, CH), 2161 (w, alkyne), 1584 (m, aromatic), 1347 (m, aryl-NH), 764 cm-1 (s, CCl3); 1H NMR (CDCl3,300 MHz, 25 °C): δ = 8.04-8.01 (m, 1H, CH), 7.87-7.83 (m, 1H, CH), 7.82-7.79 (m, 1H, CH), 7.62-7.57 (m, 1H, CH), 6.07 (br s, 1H, NH),4.58 (dd, J = 5.0 and 2.6 Hz, 2H, CH2), 2.33 (t, J = 2.56 Hz, 1H, CH); 13C NMR (CDCl3, 75.5 MHz, 25 °c): δ = 31.4 (C), 72.4 (C), 79.3 (C), 97.7 (C), 113.4 (C), 120.6 (C), 127.8 (C), 129.7 (C), 133.5 (C), 149.2 (C), 159.7 (C), 161.0 (C). HRMS-ES + : m/z [M+H]+ calcd for C12H9N3Cl3: 299.9862; found: 299.9858.
Ethan-1,2-diyl Dimethanesulfonate (9)
MsCl (7.8 mL, 101.0 mmol) was added dropwise to a solution of ethylene glycol (2.55 mL, 45.8 mmol), CH2Cl2 (45 mL) and Et3N (12.8 mL, 91.6 mmol) while under ice cooling. The cloudy white solution was stirred for1hat0°Candthen overnight at rt. The precipitate was then filtered off and the filtrate concentrated, after which it was subjected to purification on a silica gel column with 3:7 petroleum ether: EtOAc as eluent, to afford the product (9) as slightly yellow clear oil (8.94 g, 89 % yield). IR (ATR): 1352 (s, S=O), 1173 cm-1 (s, sulfonate ester); 1H NMR (CDCl3, 400 MHz, 25 °C): ö = 4.41 (s,4H, CH2), 3.04 (s, 6H, CH3); 13C NMR (CDCl3, 100 MHz, 25 °C): ö = 67.0 (2C), 35.3 (2C). The spectro-scopic data were in agreement with reported data.102-Azidoethyl Methanesulfonate (10)
Compound 9 (8.90 g, 40.8 mmol) and CH3CN (75 mL) was added to a round bottom flask. NaN3 (2.65 g, 40.8 mmol) was then added and the solution heated at refluxed (60 °C) for 22 h. The cloudy mixture was filtered and the solvent was removed from the filtrate, after which it was subjected to purification on a silica column with 3:7 EtOAc:petroleum ether to afford the desired product (10) as clear oil (3.03 g, 40 % yield). IR (ATR): 2136 (s, azide), 1347 (s, S=O), 1163 cm-1 (s, sulfonate ester); 1H NMR (cDCl3,400 MHz, 25 °c): δ = 4.29 (t, J = 4.9 Hz, 2H, CH2), 3.55 (t, J = 5.0 Hz, 2H, CH2), 3.05 (s, 3H, CH3); 13C NMR (cDCl3,100 MHz, 25 °C): δ = 66.0 (C), 48.0 (C), 35.8 (C). The spectroscopic data were in agreement with reported data.10
General Procedure for the Coupling of the Amines with 2-Azidoethyl Methanesulfonate (10)
Compound 10 (1 eq.) and Et3N (2.1 eq.) were first added to CH3CN (8 mL). The amine (2 eq.) was then added to the solution and the reaction solution was subsequently heated at reflux for 18 h under N2. The mixture was concentrated and purified on a silica column using EtOAc as the eluent, to give the corresponding products as described below.
4-(2-Azidoethyl)morpholine (11 )
Orange oil (0.376 g, 83 % yield). IR (ATR): 2093 (s, azide), 1276 cm-1 (s, ether); 1H NMR (CDCl3, 400 MHz, 25 °C): δ = 3.73-3.71 (m,4.84H, CH2), 3.35 (d, J = 6.4 Hz, 2H, CH2), 2.59 (d, J = 6.0 Hz, 2H, CH2), 2.51-2.49 (m, 4.84H, CH2); 13C NMR (CDCl3, 100 MHz, 25 °C): δ = 67.1 (C), 57.8 (2C), 53.8 (2C), 48.1 (C). The spectroscopic data were in agreement with reported data.26
1-(2-Azidoethyl)piperidine (12)
Orange yellow oil (0.352 g, 78 % yield). IR (ATR): 2096 cm-1 (s, azide); 1H NMR (CDCl3,400 MHz, 25 °C): ö = 3.28 (t, J = 6.3 Hz, 2H, CH2), 2.49 (t, J = 6.3 Hz, 2H, CH2), 2.46-2.40 (m, 4H, CH2), 1.62-1.56 (m, 4H, CH2), 1.46.-1.40 (m, 2H, CH2); 13C NMR (CDCl3, 100 MHz, 25 °C): δ = 56.7 (C), 53.6 (2C), 47.4 (C), 24.8 (2C), 23.2 (C). The spectroscopic data were in agreement with reported data.27
1-(2-Azidoethyl)pyrrolidine (13)
Orange oil (0.214 g, 63 % yield). IR (ATR): 2093 cm-1 (s, azide); 1H NMR (CDCl3, 400 MHz, 25 °C): δ = 3.40 (t, J = 6.4 Hz, 2H, CH2), 2.69 (t, J= 6.4 Hz, 2H, CH2), 2.58-2.55 (m, 4H, CH2), 1.81-1.78 (m, 4H, CH2); 13C NMR (CDCl3, 100 MHz, 25 °C): δ = 56.0 (C), 54.4 (2C), 50.4 (C), 23.7 (2C). The spectroscopic data were in agreement with reported data.28
2-Azido-N,N-dibenzylethanamine (14)
Viscous orange oil (0.316 g, 73 % yield). Mp 43 °C; IR (ATR): 2089 (s, azide), 1493 cm-1 (m, aromatic); 1H NMR (CDCl3, 300 MHz, 25 °C): ö = 7.42-7.24 (m, 10H, CH), 3.65 (s, 4H, CH2), 3.26 (t, J = 6.1 Hz, 2H, CH2), 2.71 (t, J = 6.1 Hz, 2H, CH2); 13C NMR (CDCl3, 75.5 MHz, 25 °C): ö = 138.0 (2C), 127.8 (4C), 127.3 (4C), 126.1 (2C), 57.8 (C), 52.0 (c), 48.2 (C); HRMS-ES+: m/z [M+H]+ calcd for C16H18N4: 267.161; found: 267.161.
2-Azido-N,N-diethylethanamine (15)
Yellow oil (0.198 g, 57 % yield). IR (ATR): 2092 cm-1 (s, azide); 1H NMR (CDCl3,400 MHz, 25 °C): ö = 3.29 (t, J = 6.3 Hz, 2H, CH2), 2.64 (t, J = 6.3 Hz, 2H, CH2), 2.56 (q, J = 7.1 Hz, 4H, CH2), 1.03 (t, J = 7.1 Hz, 6H, CH3); 13C NMR (CDCl3,100 MHz, 25 °C): ö = 52.3 (C), 49.7 (C), 47.4 (2C), 12.0 (2C). The spectroscopic data were in agreement with reported data.27
Phenyl Azide (17)
Aniline (0.25 mL, 2.8 mmol) was suspended in H2O (2 mL). After addition of concentrated H2SO4 (0.56 mL, 10.2 mmol) under icebath cooling and stirring, a solution of NaNO2 (0.211 g, 3.05 mmol) in H2O (1.24 mL) was added dropwise. Hexane (4 mL) was added, followed by dropwise addition of a solution of NaN3 (0.15 g, 2.8 mmol) in water (1 mL). After stirring for 2 h, the productwas extracted with CH2Cl2 (20 mL) and washed with water (20 mL). The organic layer was separated, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to give a pungent yellow liquid as product, which was used in the next step without further purification (0.33 g, 98 % yield). 1H NMR (CDCl3, 300 MHz, 25 °C): ö = 7.42-7.35 (m, 2H, CH), 7.20-7.14 (m, 1H, CH), 7.08-7.04 (m, 2H, CH); 13C NMR (CDCl3,75.5 MHz, 25 °C): ö = 140.1 (C), 129.9 (2C), 125.0 (2C), 119.2 (C). The spectroscopic data were in agreement with reported data.18
Benzyl azide (19)
To a stirred solution of benzylbromide (1.0 mL, 8.8 mmol) in a water/acetone mixture (1:4, 50 mL), NaN3 (0.856 g, 13.2 mmol) was added. The resulting suspension was stirred at RT for 24 h. CH2Cl2 (30 mL) was added to the mixture and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (3 x 10 mL) and the combined organic layers were dried over MgSO4. The solvent was removed under reduced pressure to offer the product as orange yellow pungent oil, which was used in the next step without further purification (1.030 g, 97 % yield). 1H NMR (CDCl3,300 MHz, 25 °C): δ = 7.25-7.43 (m, 5H, CH), 4.35 (s, 2H, CH2); 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 135.5 (C), 129.3 (2C), 128.3 (2C), 128.2 (C), 54.8 (C). The spectroscopic data were in agreement with reported data.19
General Procedure for the CuAAC Reaction with the Azides (11-15,17,19)
Scaffold 7 (1 eq.) and the azide (~1.1 eq.) were dissolved in CH3CN (1 mL). Et3N (1.2 eq.) was then added to the solution, followed by the addition of CuI (0.1 eq.). The mixture was then stirred for 24 h at RT or until completion by monitoring the progress of the reaction by TLC. On completion, the mixture was concentrated and diluted with DCM (20 mL), which was washed with saturated NH4Cl solution (3 x 15 mL). The organic layer was separated, dried over anhydrous MgSO4 and concentrated, after which the residue was subjected to purification on a silica column using CH2Cl2 as eluent and then with 5 % MeOH to afford the corresponding triazoles described below.
N-[(1-Benzyl-1ff-1,2,3-triazol-4-yl)methyl]-2-(trichloromethyl) quinazolin-4-amine (20)
Cream solid (0.035 g, 60 % yield). Mp 187 °C (dec); IR (ATR): 3232 (w, azole), 1259 (m, CH2), 797 cm-1 (m, CCl3); 1H NMR (CDCl3, 400 MHz, 25 °C): ö = 7.91 (dd, J = 8.4, 0.7 Hz, 1H, CH), 7.88 (d, J = 8.3 Hz, 1H, CH), 7.85 (s, 1H, CH), 7.74 (ddd, J = 8.3,7.1, 1.2 Hz, 1H, CH), 7.48-7.43 (m, 1H, CH), 7.37-7.34 (m, 3H, CH), 7.29-7.26 (m, 2H, CH), 5.48 (s, 2H, CH2), 4.93 (d, J = 5.7 Hz, 2H, CH2); 13C NMR (CDCl3,100 MHz, 25 °C): δ = 160.9 (C), 160.8 (C), 160.5 (C), 149.1 (C), 134.2 (C), 133.5 (C), 129.5 (C), 129.4 (2C), 129.2 (C), 128.7 (2C), 127.8 (C), 124.0 (C), 121.5 (C), 114.2 (c), 54.6 (C), 36.5 (C), CCl3 not detected; HRMS-ES + : m/z [M+H]+ calcd for C19H15Cl3N6: 433.0502; found: 433.0506.
N-[(1-Phenyl-1H-1,2,3-triazol-4-yl)methyl]-2-(trichloromethyl) quinazolin-4-amine (21 )
Cream solid (0.126 g, 75 % yield). Mp 195 °C (dec); IR (ATR): 3286 (w, azole), 2918 (w, CH), 1582 (m, aromatic), 1050 (m, alkene), 757 cm-1 (s, CCl3); 1H NMR (CDCl3,300 MHz, 25 °C): δ = 7.98-7.93 (m, 3H, CH), 7.79-7.70 (m, 3H, CH), 7.57-7.41 (m, 4H, CH), 5.09 (d, J = 5.7 Hz, 2H, CH2); 13C NMR (cDCl3, 75.5 MHz, 25 °C): δ = 160.8 (C), 160.5 (C), 149.1 (C), 137.1 (C), 133.5 (C), 130.0 (2C), 129.5 (C), 129.1 (C), 127.8 (C), 126.1 (C), 122.5 (C), 121.5 (C), 119.3 (2C), 114.1 (C), 35.3 (C), CCl3not detected; HRMS-ES + : m/z [M+H]+ calcd for C18H14N6Cl3: 419.0346; found: 419.0345.
N-({1-[2-(Piperidin-4-yl)ethyl]-1H-1,2,3-triazol-4-yl}methyl)-2-(trichloromethyl)quinazolin-4-amine (22)
Cream solid (0.109 g, 60 % yield). Mp 172 °C (dec); IR (ATR): 3235 (m, NH), 2923 (s, CH2), 1576 (m, aromatic), 767 cm-1 (m, CCl3); 1H NMR (CDCl3, 300 MHz, 25 °C): δ = 8.06 (s, 1H, CH), 7.93-7.91 (m, 2H, CH), 7.76-7.71 (m, 1H, CH), 7.48-7.42 (m, 1H, CH), 4.99 (d, J = 5.6 Hz, 2H, CH2), 4.48 (t, J = 6.5 Hz, 2H, CH2), 2.83 (t, J = 6.5 Hz, 2H, CH2), 2.49-2.46 (m, 4H, CH2), 1.60-1.53 (m, 4H, CH2), 1.46-1.40 (m, 2H, CH2); 13C NMR (CDCl3, 75.5 MHz, 25 °C): δ = 161.0 (C), 160.4 (C), 149.1 (C), 144.7 (C), 133.4 (C), 129.4 (C), 127.7 (C), 124.6 (C), 121.6 (C), 114.1 (C), 97.4 (C), 58.2 (c), 54.7 (2C), 47.8 (C), 36.5 (C), 25.7 (2C), 24.0 (C); HRMS-ES + : m/z [M+H]+ calcd for C19H22Cl3N7: 454.1081; found: 454.1078.
N-({1-[2-(Morpholin-4-yl)ethyl]-1H-1,2,3-triazol-4-yl}methyl)-2-(trichloromethyl)quinazolin-4-amine (23)
Cream solid (0.128 g, 70 % yield). 168 °C (dec); IR (ATR): 3227 (w, NH), 2920 (s, CH2), 1575 (w, aromatic), 765 cm-1 (w, CCl3); 1H NMR (CDCl3, 300 MHz, 25 °c): ó = 8.07 (s, 1H, CH), 7.94-7.90 (m, 2H, CH), 7.74 (ddd, J = 8.3,7.0, and 1.3 Hz, 1H, CH), 7.45 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H, CH), 4.99 (d, J = 5.8 Hz, 2H, CH2), 4.45 (t, J = 6.4 Hz, 2H, CH2), 3.67-3.64 (m, 4H, CH2), 2.81 (t, J = 6.4 Hz, 2H, CH2), 2.49-2.46 (m, 4H, CH2); 13C NMR (CDCL, 75.5 MHz, 25 °C): δ = 160.9 (C), 160.5 (C), 149.1 (C), 144.7 (C), 133.4 (C), 129.4 (C), 127.7 (C), 124.5 (C), 121.6 (C), 114.1 (C), 67.0 (2C), 58.0 (C), 53.7 (2C), 47.7 (C), 36.5 (C), CCl3 not detected; HRMS-ES + : m/z [M+H]+ calcd for C18H20Cl3N7O: 456.0873; found: 456.0875.
N-({1-[2-(Pyrrolidin-1-yl)ethyl]-1H-1,2,3-triazol-4-yl}methyl)-2-(trichloromethyl)quinazolin-4-amine (24)
Cream solid (0.119 g, 82 % yield). 155 °C (dec); IR (ATR): 3333 (m, NH), 3062 (m, CH2), 2003 cm-1 (m, azole); 1H NMR (CDCl3, 300 MHz, 25 °C): δ = 8.17 (s, 1H, CH), 7.99 (d, J = 7.9 Hz, 1H, CH), 7.88 (d, J = 8.3 Hz, 1H, CH), 7.71-7.66 (m, 1H, CH), 7.36 (t, J = 7.4 Hz, 1H, CH), 4.98 (s, 2H, CH2, 4.45 (t, J = 6.3 Hz, 2H, CH2), 2.97-2.92 (m, 2H, CH2), 2.61-2.51 (m, 4H, CH2), 1.83-1.73 (m, 4H, CH2); 13C NMR (CDCl3,75.5 MHz, 25 °C): ó = 160.9 (C), 160.5 (C), 149.0 (C), 144.7 (C), 133.2 (C), 129.1 (C), 127.5 (C), 124.6 (C), 122.0 (C), 114.2 (C), 98.6 (C), 55.6 (C), 54.2 (2C), 49.6 (C), 36.2 (C), 23.7 (2C); HRMS-ES + : m/z [M+H]+ calcd for C18H20Cl3N7: 440.0924; found: 440.0920.
N-({1-[2-(Diethylamino)ethyl]-1ii-1,2,3-triazol-4-yl}methyl)-2-(trichloromethyl)quinazolin-4-amine (25)
Cream solid (0.042 g, 52 % yield). 148 °C (dec); IR (ATR): 3333 (m, NH), 2148 (m, azole), 1965 cm-1 (m, aromatics); 1H NMR (cDCl3, 300 MHz, 25 °C): δ = 8.08 (s, 1H, CH), 7.95-7.91 (m, 2H, CH), 7.77-7.71 (m, 1H, CH), 7.48-7.42 (m, 1H, CH), 4.98 (d, J= 5.5 Hz, 2H, CH2), 4.39 (t, J = 6.4 Hz, 2H, CH2), 2.88 (t, J = 6.4 Hz, 2H, CH2), 2.54 (q, J= 7.1 Hz, 4H, CH2), 0.94 (q, J = 7.1 Hz, 6H, CH3); 13C NMR (CDCl3,75.5 MHz, 25 °c): δ = 161.0 (C), 160.5 (C), 149.1 (C), 140.2 (C), 133.4 (C), 129.4 (C), 127.6 (C), 124.8 (C), 121.7 (C), 114.2 (C), 52.9 (C), 49.2 (C), 47.5 (2C), 36.4 (c), 11.9 (2c), CCl3 not detected; HRMS-ES + : m/z [M+H]+ calcd for C18H22Cl3N7: 442.1081; found: 442.1085.
N-({1-(2-(Dibenzylamino)ethyl)-1H-1,2,3-triazol-4-yl}methyl)-2-(trichloromethyl)quinazoline-4-amine (26)
Cream solid (0.092 g, 53 % yield). 167°C (dec); IR (ATR): 3207 (s, NH), 2237 (m, azole), 1580 cm-1 (w, aromatics); 1H NMR (CDCl3, 300 MHz, 25 °C): δ = 8.00-7.97 (m, 2H, CH), 7.81-7.76 (m, 2H, CH), 7.49-7.44 (m, 1H, CH), 7.30-7.18 (m, 10H, CH), 4.98 (d, J = 5.7 Hz, 2H, CH2), 4.34 (t, J = 6.3 Hz, 2H, CH2), 3.64 (s, 4H, CH2), 2.98 (t, J = 6.3 Hz, 2H, CH2); 13C NMR (CDCl3, 75.5 MHz, 25 °c): δ = 161.5 (C), 160.4 (C), 149.1 (C), 138.7 (2C), 133.4 (C), 129.4 (C), 128.9 (4C), 128.5 (4C), 127.7 (C), 127.4 (2C), 121.6 (C), 114.7 (C), 59.1 (2C), 53.4 (C), 49.0 (C), 36.4 (C), CCl3 not detected; HRMS-ES + : m/z [M+H]+ calcd forC28H26Cl3N7:566.1394; found: 566.1391.
Acknowledgements
This work was supported by the National Research Foundation (NRF) and Stellenbosch University. We also gratefully acknowledge L. Taleli, A. Taher and D. Kleinhans for their assistance regarding this work. Finally, E. Malherbe is acknowledged for her work with the NMR spectroscopy.
References
1 A.L. Blackie, P. Beagley, S.L. Croft, H. Kendrick, J.R. Moss and K. Chibale, Bioorg. Med. Chem., 2007,15, 6510-6516. [ Links ]
2 Y. Kabri, N. Azas, A. Dumètre, S. Hutter, M. Laget, P. Verhaeghe, A. Gellis and P. Vanelle, Eur. J. Med. Chem., 2010,45, 616-622. [ Links ]
3 E.F. Elslager, P. Jacob, J. Johnson, L.M. Werbel, D.F. Worth and L. Rane, J. Med. Chem., 1978, 21, 1059-1070. [ Links ]
4 A. Mishra, K. Srivastava, R. Tripathi, S.K. Puri and S. Batra, Eur. J. Med. Chem., 2009, 44, 4404-412. [ Links ]
5 P. Verhaeghe, N. Azas, M. Gasquet, S. Hutter, C. Ducros, M. Laget, S. Rault, P. Rathelot and P. Vanelle, Bioorg. Med. Chem. Lett., 2008, 18, 396-401. [ Links ]
6 A.H. Kategaonkar, P.V. Shinde, A.H. Kategaonkar, S.K. Pasale, B.B. Shingate and M.S. Shingare, Eur. J. Med. Chem., 2010, 45, 3142-3146. [ Links ]
7 Y.-C. Duan, Y.-C. Ma, E. Zhang, X.-J. Shi, M.-M. Wang, X.-W. Ye and H.-M. Liu, Eur. J. Med. Chem., 2013, 62, 11-19. [ Links ]
8 M. Meldal and C.W. Torn0e, Chem. Rev., 2008,108, 2952-3015. [ Links ]
9 J.E.MosesandA.D. Moorhouse, Chem. Soc. Rev., 2007,36,1249-1262. [ Links ]
10 H.H. Kinfe and Y.H. Belay, S. Afr. J. Chem., 2013, 66, 130-135. [ Links ]
11 A. Sugawara, T. Tanaka, T. Hirose, A. Ishiyama, M. Iwatsuki, Y. Takahashi, K. Otoguro, S. Ōmura and T. Sunazuka, Bioorg. Med. Chem. Lett, 2013, 23, 2302-2305. [ Links ]
12 E.M. Guantai, K. Ncokazi, T.J. Egan, J. Gut, P.J. Rosenthal, P.J. Smith and K. Chibale, Bioorg. Med. Chem., 2010,18, 8243-8256. [ Links ]
13 P. Singh, P. Singh, M. Kumar, J. Gut, P. J. Rosenthal, K. Kumar, V Kumar, M.P. Mahajan and K. Bisetty, Bioorg. Med. Chem. Lett, 2012, 22, 57-61. [ Links ]
14 R. Raj, P. Singh, P. Singh, J. Gut, P. J. Rosenthal and V. Kumar, Eur. J. Med. Chem., 2013, 62, 590-596. [ Links ]
15 P. Verhaeghe, P. Rathelot, A. Gellis, S. Rault and P. Vanelle, Tetrahedron, 2006, 62, 8173-8176. [ Links ]
16 P. Verhaeghe, N. Azas, S. Hutter, C. Castera-Ducros, M. Laget, A. Dumètre, M. Gasquet, J.-P. Reboul, S. Rault, P. Rathelot and P. Vanelle, Bioorg. Med. Chem., 2009,17, 4313-322. [ Links ]
17 C. Tahtaoui, I. Parrot, P. Klotz, F. Guillier, J.-L. Galzi, M. Hibert and B. Ilien, J. Med. Chem., 2004,47, 4300-315. [ Links ]
18 A. Cwiklicki and K. Rehse, Arch. Pharm. Pharm. Med. Chem, 2004, 337, 156-163. [ Links ]
19 L.S. Campbell-Verduyn, L. Mirfeizi, R.A. Dierckx, P.H. Elsinga and B.L. Feringa, Chem. Commun., 2009, 0, 2139-2141. [ Links ]
20 K. Yearick, K. Ekoue-Kovi, D.P. Iwaniuk, J.K. Natarajan, J. Alumasa, A.C. de Dios, P.D. Roepe and C. Wolf, J. Med. Chem., 2008, 51, 1995-1998. [ Links ]
21 J.-F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev, 2008,60,958-970. [ Links ]
22 M. Dabiri, P. Salehi, M. Bahramnejad and F. Sherafat, Comb. Sci., 2010, 12, 638-642. [ Links ]
23 S. Bernard, D. Defoy, Y. L. Dory and K. Klarskov, Bioorg. Med. Chem. Lett., 2009,19, 6127-6130. [ Links ]
24 W. Trager and J.B. Jensen, Science, 1976,193, 673-675. [ Links ]
25 M.T. Makler, J.M. Ries, J.A. Williams, J.E. Bancroft, R.C. Piper, B.L. Gibbins and D.J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 739-741. [ Links ]
26 T. Suzuki, Y. Ota, M. Ri, M. Bando, A. Gotoh, Y. Itoh, H. Tsumoto, P.R. Tatum, T. Mizukami, H. Nakagawa, S. Iida, R. Ueda, K. Shirahige and N. Miyata, J. Med. Chem., 2012, 55, 9562-9575. [ Links ]
27 L. Le Corre, A.-L. Girard, J. Aubertin, F. Radvanyi, C. Benoist-Lasselin, A. Jonquoy, E. Mugniery, L. Legeai-Mallet, P. Busca and Y. Le Merrer, Org. Biomol. Chem., 2010, 8, 2164-2173. [ Links ]
28 S. Wang, Q. Wang, Y. Wang, L. Liu, X. Weng, G. Li, X. Zhang and X. Zhou, Bioorg. Med. Chem. Lett, 2008,18, 6505-6508. [ Links ]
 Correspondence:
Correspondence:
E-mail: mblackie@sun.ac.za
Received 25 April 2013
Revised 10 June 2013
Accepted 22 June 2013


{kind=link}
{kind=link}