










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban ago. 2013
RESEARCH ARTICLE
Hydrogenation of cinnamaldehyde over an ionic cobalt promoted alumina-supported palladium catalyst
Elizabeth Erasmus*
Chemistry Department, University of the Free State, P.O. Box 319, Bloemfontein, 9300, South Africa
ABSTRACT
Nano γ-alumina-supported transition metal-based mono- (Pd0/γ-Al2O3 or CoIII/γ-Al2O3) and bimetallic catalysts (Pd0CoIII/γ-Al2O3), were prepared by wet impregnation, oxidation and in situ reduction of the palladium. The palladium(II) acetate [PdII3(OOCCH3)6], cobalt(II) acetate [CoII(OOCCH3)2].4H2O and palladium, cobalt bridging tetraacetate [PdIICoII(OOCCH3)4].H2O.2CH3COOH, were used as catalyst precursors to prepare a 7.4 % mol catalyst/support loading. The oxidation states of these activated catalysts were studied by XPS. These catalysts were evaluated for hydrogenation of cinnamaldehyde in a biphasic reaction under 3 MPa of hydrogen at 80 °C. The Pd0/γ-Al2O3 selectively catalyzed the hydrogenation of C=C bond in cinnamaldehyde to form phenylpropanol (100 %). In contrast, catalysis with Pd0CoIII/γ-Al2O3 hydrogenated both C=C and C=O bonds consecutively, forming 65 % phenylpropanal and 35 % phenylpropanol with TOF = 47.2 h-1. No reaction occurred when CoIII/γ-Al2O3 was used.
Keywords: Bimetallic heterogeneous catalyst, palladium, cobalt, hydrogenation, unsaturated aldehydes.
1. Introduction
The catalytic hydrogenation of α,β-unsaturated aldehydes is a useful transformation in organic synthesis to produce fine chemicals.1 To selectively hydrogenate carbon-carbon double bonds (C=C) in molecules containing other functional groups like aldehydes is a very important process in the fragrance, flavouring and pharmaceutical industry.2-5 In α,β-unsaturated aldehydes the reduction of the carbonyl group (C=O) is in competition with the hydrogenation of the C=C bond, where the hydrogenation of the C=C bond is thermodynamically more favoured by about 35 kJ/mol.6 Consecutive hydrogenation can also occur first, leading to the formation of the saturated aldehyde followed by the formation of the saturated alcohol, or hydrogenation to form the unsaturated alcohol and then reduction to form the saturated alcohol. Depending on the reaction conditions and catalyst type, hydrogenation can be selective for a specific functional group or it can proceed in steps to form the fully hydrogenated product.
Cinnamaldehyde is one of the most popular substrates to test catalytic performance of a hydrogenation catalyst since two different unsaturated bonds (C=C and C=O) are present in the same molecule.7-10 The hydrogenation of cinnamaldehyde can proceed via two routes as shown in Fig. 1, depending on the catalyst and reaction conditions. The different products that can form are hydrocinnamaldehyde, cinnamyl alcohol and phenyl propanol.

Palladium is well known to be very active for C=C bond hydrogenation,11-13 and in the hydrogenation of unsaturated aldehydes it gives almost 100 % yield in saturated aldehyde.8 The selectivity of Pd towards the formation of saturated carbonyls as products, is explained by the preferred adsorption of the C=C bond on Pd.14
It has been shown that promotion of metallic catalysts with an ionic compound increase the hydrogenation rate of theC=O bond of unsaturated aldehydes,15 due to the activation of the C=O bond by the ionic species.
This article reports on the preparation and characterization of an ionic Co(III)-promoted Pd0/Al2O3 catalyst, using the organo-metallic single-source catalyst precursor [PdIICoII(µ-OOCCH3)4]. H2O.2CH3COOH (1). The catalytic activity for the hydrogena-tion of cinnamaldehyde to hydrocinnamaldehyde and phenyl propanol under mild conditions, i.e. 80 °C and 3 MPa H2 were investigated. Comparison was made with the unpromoted Pd0/Al2O3 catalyst and the promoted catalyst Pd0CoIII/Al2O3 catalyst prepared by 1 and the physical mixing of the two mono-metallic acetate precursors, [(Pd3(OOCCH3)6](2)and [Co(OOCCH3)2].4H2O (3).
2. Experimental
2.1. Chemicals
Ethanol, palladium(II) chloride, cobalt(II) acetate tetrahydrate were purchased from Aldrich and used without any further purification. Catalyst support used was nano γ-Al2O3 with a surface area of 220 m2 g-1. The attenuated total reflectance, ATR FTIR spectra were recorded on a Nexus FTIR equipped with a Nicolet Smart Golden Gate. XPS data were recorded on a Kratos AXIS Ultra spectrometer, equipped with a monochromatic Al Kα X-ray source and a delay-line detector. Spectra were obtained using the aluminium anode (Al Kα = 1486.6 e V) operating at 150 W, with the survey scans a constant pass energy of 160 eV was used and with the region scans a constant pass energy of 40 e V was used. The background pressure was 2 x 10-9 mbar and data was fitted on CasaXPS. Temperature programmed reduction (TPR) experiments was performed by measuring the H2 consumption of an oxidized PdIVCoIII/Al2O3 catalyst as a function of temperature. The sample was heated at a rate of 5 K min-1 to a temperature of 973 K in a 10 mL min-1 H2/N2 gas flow (4% H2 in N2). Tripalladium(II) hexaaacetate [(PdII3(OOCCH3)6], 2, was prepared according to published procedures.16
2.2. Preparations
2.2.1. Palladium-Cobalt Bridging Tetracarboxylate Paddlewheel [Pd IICoII(COOCH3)4].2H2O.CH3COOH, 1
[PdIICoII (COOCH3)4].H2O.2CH3COOH was prepared according to published procedures.17,18
A mixture of 2 (340 m, 0.5 mmol) and 3 (362 mg, 1.45 mmol) in 10 mL acetic acid was refluxed for 2 h. The mixture was left to cool slowly, overnight. Brown precipitate was filtered off and washed with acetic acid and water and dried. Yield 710 mg (1.43 mmol, 98 %). IR (cm-1): 1699 (νas COO), 1610 νas COO), 1508 (νas COO), 1419 (νs COO), 1377 (νs COO), 1325 (νs COO).). The crystal structure of this compound is reported elsewhere.17
2.2.2. Incipient Wet Impregnation of Nano γ-Al2O3 with 1, 2 and 3
1 (or 2 and/or 3) (0.6 mmol) dissolved in acetone was slowly added to nano γ-Al2O3 (830 mg, 8.14 mmol), the mixture was stirred for 24 h and the solvent was removed under vacuo to yield a catalyst loading of 21-22 molecules nm-2 or 3.6 (± 0.1) x 10-5 mol m-2 or 7.4 % mol catalyst mol-1 solid support.
2.2.3. Oxidation of 1, 2 and/or 3 on Nano γ-Al2O3
Oxidation of the freshly prepared 1 or 2 and/or 3/nano γ-Al2O3 was achieved by placing the sample in an oven. It was then oxidized under O2/Ar flow of 250 mL min-1, while increasing the temperature from 25 °C to 200 °C at a rate of 10 °C min-1, the sample was then kept at this temperature for 30 min before the temperature was further increased at a rate of 10 °C min-1 to 500 °C. The sample was then kept at this temperature for 4 h before it was allowed to cool spontaneously overnight. Oxidation of sample prepared 1/nano γ-Al2O3 produced PdIVCoIII/γ-Al2O3, 4.
2.2.4. In situ Reduction of the Pd
The oxidized catalyst was loaded into a 50 mL self-made reactor. After the reactor was flushed three times with H2 and heated to a temperature of 80 °C, the H2 pressure was increased to 3 MPa and the reduction of Pd with H2in situ was allowed to proceed for 2 h. ln situ reduction of the Pd produced Pd0CoIII/ γ-Al2O3, 5, (prepared by 1), Pd0+CoIII/γ-Al2O3, 6 (prepared by physically mixing 2 and 3 in the same solution), Pd0/γ-Al2O3, 7 (prepared by 2) and CoIII/γ-Al2O3, 8 (prepared by 3).
2.2.5. Hydrogenation of Cinnamaldehyde with Pd0CoIII or Pd0 or CoIII/γ-Al2O3 Catalyst
In a typical experiment, after loading of the catalyst PdIVCoIII or PdIV or CoIII/γ-Al2O3. (30 mg) and in situ reduction of palladium (Pd0CoIII or Pd0/γ-Al2O3), cinnamaldehyde (3 mL, 23.8 mmol) was added to the 50 mL self-made reactor. The reaction was allowed to proceed for 24 h at a temperature of 80 °C, and H2 pressure of 3 MPa.
Turnover frequency was calculated by:
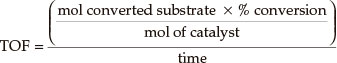
3. Results and Discussion
The heterobimetallic compound 1, of the form [PdIICoII (COOCH3)4].H2O.2CH3COOH, was prepared by reacting 2 with the 3, in glacial acetic acid to yield the desired product in almost quantitative yield (98 %). Water is an axial ligand, associated with the Co via hydrogen interaction. There are also two acetic acid molecules associated between two of the [PdIICoII(COOCH3)4] complexes, which can been seen in the ATR FTIR of the compound (Fig. 2). Since this compound is known and its crystal structure was already determined,17 comparison of IR data confirmed that the correct product was obtained.

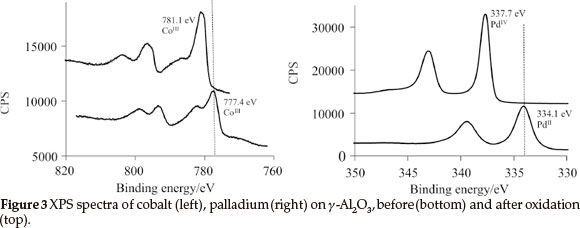
1, 2, 3 and the physically mixed solution of 2 and 3 were deposited onto the γ-Al2O3 to produce mono- and bimetallic heterogeneous catalysts by means of wet impregnation followed by oxidation. XPS results of the oxidized samples revealed that PdIV (3d5/2 = 337.7 eV) and CoIII (2p3/2 = 781.1 eV) was present on the surface (see Fig. 3). These binding energies are within experimental error the same that was obtained for 1 impregnated by means of spin coating onto a Si(OH)/SiO2/Si(100) two dimensional wafer.18
A temperature programmed reduced (TPR) study was performed on 4, [PdIVCoIII/γ-Al2O3] and Fig. 4 shows the rate of consumption of hydrogen on 4. Three reduction peaks were observed, a sharp, well-defined peak at 82 °C and two large broad peaks at 121 °C and 542 °C. The TPR peak at 82 °C corresponds to the reduction of Pd, it is at a slightly higher temperature than reported values for Pd on alumina, 67 °C19 and 73 °C.20 This change is possibly due to the presence of Co and the acetate ligands, which shields the Pd, making H2 consumption more difficult. The flat broad peak at 121 °C is probably due to the consumption of H2 by the organic ligand, the other larger broad peak at 542.9 °C is due to H2 consumption on Co. Literature reports that Co on alumina shows two sharp, well-defined TPR peaks at 549 °C and 635 °C,21 which is in contrast to what was found for the Co in 4. This could be indicative of Pd-promoted reduction of the Co as well as the broad shape of the peak shows the presence of highly dispersed Co-containing particles on the surface.22 This shows that the single source precursor approach yield good dispersion of the metals.
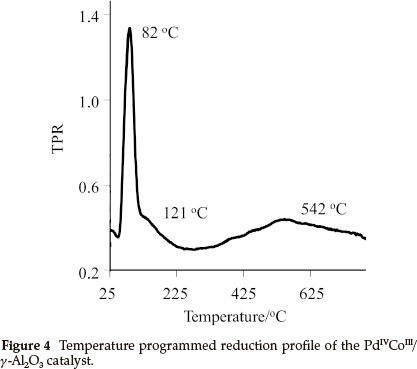
From the data obtained from the TPR, which shows that Pd in 4, has a reduction maximum at 82 °C in a 10 mL min-1 H2/N2 gas flow. It can be assumed that the Pd in 4 is fully reduced in situ at 80 °C and 3 MPa H2, which implies that the active catalyst is Pd0CoIII/Al203,5.
In previous work 1 was used as a single source catalyst precursor to prepare the two dimensional model catalyst PdIVCoIII/Si(0H)/ Si02/Si(100), which was found to work very well for aerobic oxidation of alcohols.18 The purpose of this study is to see if using 1 as a single source catalyst precursor could work as a dual catalyst. 5, prepared from 1 and the physically mixed Pd0+CoIII/Al203, 6, prepared from 2 and 3, were applied as catalysts for hydrogenation of cinnamaldehyde, under solvent-free conditions at 3 MPa pressure of H2 and temperature of 80 °C. Parallel control reactions catalyzed by Pd0/Al203, 7, and CoIII/Al203, 8, were also carried out.
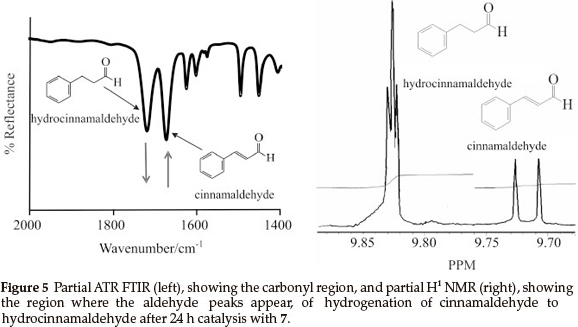
For comparison, samples of the reduction reactions were taken after 24 h and were analyzed by means of ATR FTIR and H1 NMR, as an example the spectra obtained by 7 is shown in Fig. 5. On the ATR FTIR (Fig. 5, left), the disappearance of the CO stretching peak of the cinnamaldehyde (1668 cm-1) and the appearance of the CO stretching of hydrocinnamaldehyde (1720 cm-1) was followed. Conversion after 24 h (Table 1) was calculated by the ratio of the aldehyde peaks (see H1 NMR in Fig. 5 , right), cinnamaldehyde appears at 9.71 ppm as a doublet and hydrocinnamaldehyde appears at 9.83 ppm as a triplet.

The ATR FTIR graphs of all the tested catalysts after 24 h catalytic hydrogenation of cinnamaldehyde at 80 °C and 3 MPa H2 pressure is shown in Fig. 6. It can be concluded that CoIII/Al203 is catalytically inactive towards hydrogenation since no conversion was found. After 24 h, 7 converted 82 % of cinnamaldhyde to hydrocinnamaldehyde and it was found that after 72 h there was 100 % conversion, with no further conversion to phenyl-propanol and a TOF = 38.7 h-1. 6 showed a conversion of 58 % and a TOF = 27.5 h-1 after 24 h, which is considerably lower that was found for 7. This shows that the addition of CoIII to Pd0 lowers its activity towards conversion of cinnamaldehyde to hydrocinnamaldehyde. This probably due to aggregation and segregation, with the CoIII only acting as a spectator and being positioned on the outside of the particle, thereby lower the 'active' surface of the Pd, making the conversion slower. However, 5 showed that after 24 h no cinnamaldehyde was present with a TOF = 47.2 h-1. A mixture of products was obtained, 35 % hydrocinnamaldehyde and 65 % phenylpropanol. Even after 72 h this ratio of hydrocinnamaldehyde to phenylpropanol did not change. This implies that after 24 h, the catalysis was complete or that the catalyst got poisoned. This result is in contrast to what was observed when a different support and physical mixture of Pt and Co is used, PtCo/ZrO results in 93.8 % conversion of cinnamaldehyde to cinnamaylalcohol.23 It was also shown that when Cu on silica support is promoted with Pd, the reaction is accelerated but the selectivity towards cinna-maylalcohol is lost, similar to what we observed.24

When a single source catalyst precursor is used, a more homogeneous dispersion of the two metals is obtained thus the addition of the CoIII to Pd0 not only increase the conversion rate of cinnamaldehyde to hydrocinnamaldehyde but also facilitates the hydrogenation of hydrocinnamaldehyde to phenylpropanol. This confirms that the addition of an ionic species, in this case CoIII, to the metallic Pd catalyst activates the hydrogenation of the C=O bond, after hydrogenation of the C=C bond is complete.
Figure 7 shows the ATR FTIR graphs of the catalytic reaction with 5 at certain time intervals. It can be clearly seen how the CO stretching peak of the cinnamaldehyde (1668 cm-1) disappears and the CO stretching hydrocinnamaldehyde (1720 cm-1) appearance, which is accompanied by the formation of CH stretching at 2919 cm-1. In the last spectra after 72 h, the appearance of the OH stretching (3403 cm-1) and the CO stretching peak (1676 cm-1) of the phenylpropanol is observed.

Since catalyst 6, which was prepared by physically mixing 2 and 3, showed a decrease in catalytic hydrogenation of cinnamaldehyde and 5 catalyzed the reaction to 65 % phenylpropanol, it can be concluded that when using the organometallic precursor, 1, a catalytic synergism is obtained due to a special interaction either between Pd0 and CoIII or between the metals and the solid support.
Under the solvent free condition used for preliminary selective hydrogentation of the cinnamaldehyde to phenylpropanol, the catalyst needs regeneration by oxidation and in situ reduction of the Pd to reuse the catalyst.
4. Conclusion
The single source precursor [PdIICoII(COOCH3)4].H2O. 2CH3COOH, 1, was successfully used to impregnate nano γ-Al2O3 to form a bimetallic catalyst. The XPS of the oxidized catalyst showed that the metals oxidation states are PdIV and CoIII. TPR studies revealed that the PdIV get reduced in situ at 80 °C and 3 MPa pressure of H2 to yield an active catalyst with a form of Pd0CoIII/Al2O3, 5. The hydrogenation of cinnamaldehyde revealed that the catalyst obtained by physically mixing 2 and 3 to give Pd0+CoIII/Al2O3, 6, showed a lower conversion than the Pd0/Al2O3, implying that the addition of CoIII to Pd0 lowers its hydrogenation activity, most probably by lowering the active catalyst surface area. Catalyst 5, prepared from the single source catalyst precursor 1, revealed that hydrogenation of both C = C and C=O bond occurs and the product ratio obtained is 35 % hydrocinnamaldehyde and 65 % phenylpropanol.
Acknowledgements
The author acknowledges financial support from SASOL and the UFS during the course of this study. Tiny Verhoeven is acknowledged for recording of the XPS data.
References
1 M. Boudart, Nature, 1994, 372, 320-320. [ Links ]
2 H.U. Blaser, Catal. Today, 2000, 60, 161-165. [ Links ]
3 S. Bailey and F. King, in Fine Chemicals through Heterogeneous Catalysis, (R.A. Sheldon and H. van Bekkum, eds.), 1st edn., Wiley-VCH, Weinheim, Germany, 2001. [ Links ]
4 H.U. Blaser, F. Spindler and M. Studer, Appl. Catal. A, 2001, 221, 119-143. [ Links ]
5 P. Gallezot and D. Richard, Catal. Rev. Sci. Eng., 1998, 40, 81-126. [ Links ]
6 C. Mohr and P. Claus, Sci. Progress, 2001, 84, 311-334. [ Links ]
7 A. Corma, H. Garcia and A. Leyva, J. Mol. Catal. A. Chem, 2005, 230, 97-105. [ Links ]
8 J.P Tessonnier, L. Pesant, G. Ehret, M.J. Ledoux and C. Pham-Huu, Appl. Catal. A: Gen, 2005, 288, 203-210. [ Links ]
9 J. Qiu, H. Zhang, X. Wang, H. Han, C. Liang and C. Li, React. Kin. Catal. Lett., 2006, 88, 269-275. [ Links ]
10 H. Wang, Y. Shu, M. Zheng and T. Zhang, Catal. Lett., 2008, 124, 219-222. [ Links ]
11 C. Pham-Huu, N. Keller, G. Ehret, L.J. Charbonniere, R. Ziessel and M.J. Ledoux, J. Mol. Catal. A: Chem, 2001, 170, 155-163. [ Links ]
12 M. H. Chen, W. Chu, X. Y. Dai and X. W Zhang, Catal. Today, 2004, 89, 201-204. [ Links ]
13 T. Maegawa, T. Takahashi, M. Yoshimura, H. Suzuka, Y. Monguchi and H. Sajiki, Adv. Synth. Catal, 2009, 351, 2091-2095. [ Links ]
14 F. Delbecq and P. Sautet, J. Catal, 1995, 152, 217-236. [ Links ]
15 V. Ponec, App. Catal. A: General, 1997, 149, 27-48. [ Links ]
16 T.A. Stephenson, S.M. Morehouse, A.R. Powell, J.P. Heffer and G. Wilkinson, J. Chem. Soc, 1965, 3632-3640. [ Links ]
17 N.S. Akhmadullina, N.V. Cherkashina, N.Y. Kozitsyna, I.P. Stolarov, E.V. Perova, A.E. Gekhman, S.E. Nefedov and M.N. Vargaftik, I.I. Moiseev, Inorg. Chim. Acta, 2009, 362, 1943-1951. [ Links ]
18 E. Erasmus, J.C. Swarts and J.W. Niemantsverdriet, Langmuir, 2012, 28, 16477-16484. [ Links ]
19 V.V. Alegre, M.A.P. da Silva and M. Schmal, Catal. Comm., 2006, 7, 314-322. [ Links ]
20 M. Conceição Greca, C. Moraes and A.M. Segadães, Appl. Catal. A, 2001, 216, 267-276. [ Links ]
21 A.M. Hilmen, D. Schanke and A. Holmen, Catal. Lett., 1996, 38, 143-147. [ Links ]
22 L. Guczi, Z. Schay, G. Stefler and F. Mizukami, J. Mol. Catal. A: Chem., 1999, 141, 177-185. [ Links ]
23 G.H. Lai, R.X. Zhou, X.X. Han and X.M. Zheng, Chin. Chem. Lett., 2004, 15, 1494-1496. [ Links ]
24 A. Chambers, S.D. Jackson, D. Stirling and G. Webbz, J. Catal., 1997, 168, 301-314. [ Links ]
Received 20 September 2011
Revised 16 August 2012
Accepted 22 April 2013
* To whom correspondence should be addressed. E-mail: erasmuse@ufs.ac.za

