










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
RESEARCH ARTICLE
Intermolecular interactions and thermodynamic properties of 3,6-diamino-1,2,4,5-tetrazine-1,4-dioxide dimers: A density functional theoretical study
Ning Ning ZhaoI; Ya Ling ZhaoI; Yin HuI, II; Hai Xia MaI, *; Feng Qi ZhaoII; Ji Rong SongI, III
ISchool of Chemical Engineering/Shaanxi Key Laboratory of Physico-Inorganic Chemistry, Northwest University, Xi'an, 710069, China
IIScience and Technology on Combustion and Explosion Laboratory, Xi'an Modern Chemistry Research Institute, Xi'an, 710065, China
IIIDepartment of Conservation Technology, The Palace Museum, Beijing, 100009, China
ABSTRACT
Three fully optimized structures of 3,6-diamino-1,2,4,5-tetrazine-1,4-dioxide (LAX-112) dimers have been obtained with the density functional theory (DFT) method at the B3LYP/6-311++G level. Vibrational frequency calculations were carried out to ascertain that each structure is a minimum (no imaginary frequencies). The intermolecular interaction energy is calculated with the basis set superposition error (BSSE) correction and zero point energy (ZPE) correction. The greatest corrected binding energy among the three dimers is -42.38 kJ mol-1. The charge redistribution mainly occurs on the adjacent O(N)......H atoms between submolecules and the charge transfer between two subsystems is very small. Natural bond orbital (NBO) analysis was performed to reveal the origin of the interaction. Based on the vibrational analysis, the standard thermodynamic functions (heat capacities (cop), entropies (S°m) and enthalpies (H°m)) and the changes of thermodynamic properties from the monomer to dimer with the temperature ranging from 200.00 K to 800.00 K have been obtained using statistical thermodynamics. The results show that the strong hydrogen bonds dominantly contribute to the dimers, while the bonding energies are not only determined by the hydrogen bonding. The dimerization process of dimer II can occur spontaneously at room temperature.
Keywords: 3,6-Diamino-1,2,4,5-tetrazine-1,4-dioxide (LAX-112), intermolecular interaction, density functional theory (DFT), natural bond orbital (NBO) analysis, thermodynamic properties.
1. Introduction
Unlike the traditional energetic compounds, such as 2,4,6-trinitrotoluene (TNT) and hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX), which derive their energies from the oxidation of the carbon and hydrogen atoms in the molecules,1 high-nitrogen heterocyclic materials (tetrazines, furazans, tetrazoles, etc.) typically have large positive heats of formation as their source of energy. They consist of a large number of energetic N-N and C-N bonds in their structures, and the contents of high nitrogen and low carbon hydrogen make them easily obtain oxygen balance. In addition, this kind of materials tends to have an additional benefit of being less toxic to users and the environment.2 Tetrazine compounds are the typical high-nitrogen materials with a nitrogen content of 68.3 % in the tetrazine ring which is an effective structure unit in designing high energetic materials (HEMs). This kind of materials has a great deal of potential applications in the insensitive explosives, low signature propel-lants, gas generants and low-smoke pyrotechnics.3-7 Owing to the gas production from decomposition in most cases, tetrazine compounds were considered as a potential formula in the eco-friendly pyrotechnics.8
EMs are an aggregative and mixed system and the study on intermolecular interactions of EMs has attracted a wide attention. Intermolecular forces have significant effects on many physical properties of EMs, such as diffusion, aggregation, density and detonation. In addition, intermolecular interactions are also closely related with safety and mechanical properties of EMs. Therefore, there is an important academic and application significance to carry out studies on the intermolecular interactions of EMs. In recent years, the intermolecular interactions in a series of explosives have obtained some meaningful informa-tion9-18 which is valuable for further study of EMs. The inter-molecular interactions of 3,6-dihydrazino-1,2,4,5-tetrazine (DHT)19 and 3,6-Diamino-1,2,4,5-tetrazine (DATZ)20 were examined, which was found that the strong hydrogen bonds domi-nantly contribute to the dimers.
3,6-diamino-1,2,4,5-tetrazine-1,4-dioxide (LAX-112) is one of the early used insensitive tetrazine explosives21-30 with its nitrogen content, enthalpy of formation, density, critical diameter of 58.3 %, 164 kJ mol-1, 1.834 gcm-3, and less than 6 mm respectively. Its detonation velocity and detonation pressure are 8.26 km s-1 and 24.2 GPa respectively. The enthalpy of formation, detonation velocity and detonation pressure of TATB (1,3,5-triamino-2,4,6-trinitrobenzene) are 140 kJ mol-1, 7.62 km s-1 and 25.9 GPa respectively. Therefore, LAX-112 has a better explosion performance than TATB.22,31-34 The synthesis, thermal behaviour on LAX-112 were reported,21-30. In this paper, we theoretically investigated the intermolecular interaction and thermodynamic properties of LAX-112 dimers.
2. Computational Methods
The geometries of LAX-112 monomer and all its possible stable dimers as obtained using the Chem3D software package were fully optimized by the Berny method35 at the DFT(B3LYP) level with 6-311 + + G**. Single point energy calculations were also performed at the MP2/6-311 + + G** and B3LYP/aug-cc-pVDZ levels on the dimer structures obtained at the B3LYP/6-311 + +G** level. Natural bond orbital analysis and frequency calculations were carried out on each optimized structure. Thermodynamic data and their changes upon dimerizing were derived from statistical thermodynamics36 based on the frequency calculation. The intermolecular interaction energies of the studied dimers were corrected both with basis set superposition error (BSSE) and zero point energies (ZPE). All these calculations have been carried out with the Gaussian 9837 program using the default Gaussian convergence criteria.
It has been suggested that the M06-2X, M05-2X, M06-HF, and M06 functionals are the best functionals for the study of non-covalent interactions38, therefore, the geometries of the monomer and the three dimmers were also fully optimized at M06-2X/6-311 + +G** by Gaussion 0939. The interaction energies were compared with the above methods.
The intermolecular interaction energies of (LAX-112)2 were evaluated from the energy difference between the dimer and monomer (Eq. 1).

where Ea(b) and Ea-b are the total energies of the monomer and dimer (LAX-112)2, respectively. The zero point energy corrections (ZPEC) are adopted in the present work based on Eq. 2:
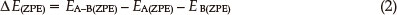
where Ea(b)(zpe) and Ea-b(zpe) are the zero point energies of the monomer and dimer (LAX-112)2, respectively. To correct for the basis set superposition error, the Boys and Bernardi's counterpoise procedure (CP)40-42 is employed as follows:
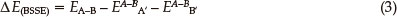
where EA-Ba'(b') is the calculated energy of the LAX-112 monomer A or B with its geometry in (LAX-112)2 but using the basis functions of the full dimer. Finally the corrected interaction energies ΔEcorr (Eq. 4) were calculated by addition of ΔE(ZPE) and ΔE(BSSE) to the interaction energy term (Eq.(1))
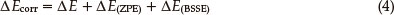
3. Results and Discussion
3.1 Optimized Structures
Three stable structures of LAX-112 dimers are presented in Fig. 1.
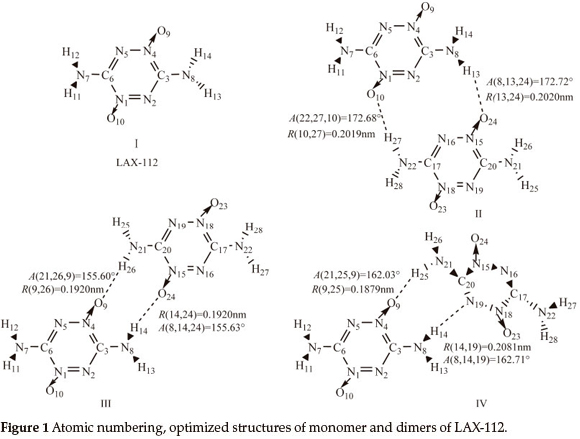
In the case of dimers II-IV, each submolecule consists of one donor atom and one acceptor atom. Dimers II and III are symmetrical and dimer IV is unsymmetrical. There are two hydrogen bonding interactions of N22-H27...O10 and N8-H13 ...O24 in dimer II. In dimer III, hydrogen bonding interactions are N21-H26...O9 and N8-H14...O24 which consist of a ten-numbered ring. The H-bonding lengths usually determine the binding energies when the intermolecular contacts are similar. Judged by the intermolecular distance, as shown in Figure 1, the strength of interactions maybe in the order: III>II.
Some geometrical parameters are collected in Table 1. The monomer and the dimers differ in the following aspects: (a) For dimer II, the lengths of N1-O10 and N15-O24 increase by 2.0 pm, the N8-H13 and N22-H27 lengths increase by 1.5 pm, the N4-C3 and N18-C17 increase by 1.9 pm, while the N1-C6 and N15-C20 lengths decrease by 1.8 pm, the N8-C3 and N22-C17 lengths decrease by 1.2 pm. (b) For dimer IV, the lengths of N4-O9, N18-O23, N21-H25, N2-C3 and N15-C20 in increase by 1.0 pm, 0.7 pm, 1.1 pm, 1.0 pm and 1.3 pm, respectively, while N8-C3, N21-C20 and N18-C17 decrease by 1.3 pm, 1.5 pm and 1.2 pm, respectively. (c) For dimer III, the bond lengths change less than those of the other two dimers. The lengths of N4-O9, N15-O24, N8-H14 and N21-H26 increase by 0.8 pm, the N2-C3 and N19-C20 lengths increase by 0.9 pm, while the lengths of N8-C3 and N21-C20 decrease by 0.8 pm. (d) The changes of angles are within 4.6 ° and those of the dihedrals are small (the data was omitted in the table) indicating that the intermolecular interactions have little influence on angle-torsion of the monomer and inner rotation of a single bond.
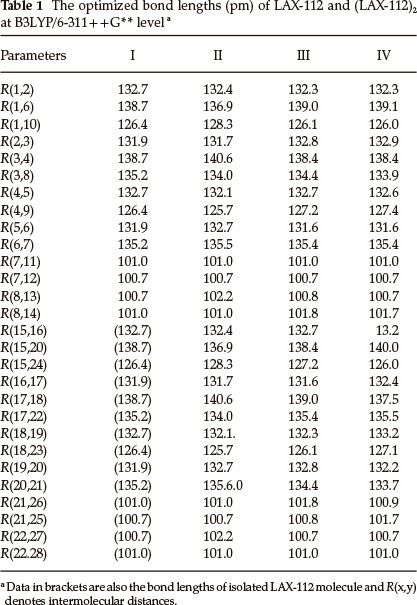
3.2 Binding Energies
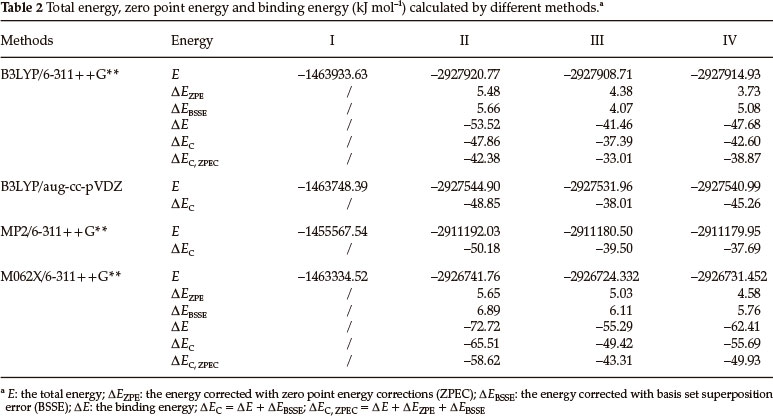
Table 2 shows both the uncorrected and corrected binding energies. There are no imaginary frequencies for any of the structures in Table 2, indicating that the structures in Figure 1 are indeed the minima on their potential energy surfaces.
To determine the appropriateness of the chosen basis set (6-311 + +G**) for the calculations, we have also determined the binding energies with the aug-cc-pVDz basis set. The maximum differences in the values of (ΔE)C caused by using the two basis sets are 2.66 kJ mol-1. The corrected binding energies in Table 2 give the same stability order for the dimers with 6-311 + +G** and aug-cc-pVDz, suggesting that the energies are close to the basis set limit. Furthermore, to confirm the reliability of the calculated results, single point energies were calculated with the MP2 method using the B3LYP/6-311++G** optimized structures. The differences in values of (ΔE)C caused by using these two methods are not large. However, the order of (ΔE)C is different. For the H-bonded (LAX-112)2 the values of (ΔE)C calculated using B3LYP/aug-cc-pVDz are relatively close to those using B3LYP/6-311 + +G**. Moreover, when the intermolecular interactions are strong, it is very reliable to calculate binding energies by using the DFT method. The calculated intermolecular interaction energies with MP2 are usually larger than those using B3LYP and the same basis set.43 On the contrary, the calculated energy for structure IV with MP2/6-311 + +G** is lower than that with B3LYP. The calculated energies for the three dimers at the M062X/6-311 + +G** level are a little higher than those at B3LYP/6-311++G**, however the stability order at these two levels are the same. Therefore, the interaction energies calcu- lated with B3LYP is reasonably reliable for these H-bonded complexes. (Our discussion is still based on the results using B3LYP/6-311++G**.)
The corrected binding energy for dimer II is -42.38 kJ mol-1, the binding energy for each hydrogen bond is -21.19 kJ mol-1. This value is larger than the best experimentally estimated dissociation energy (15 kJ mol-1) of the water dimer,44 indicating that the hydrogen bonding is strong. The binding energy for dimer III is the lowest among the three dimers (-33.01 kJ mol-1), showing that the N(N19) atom in N→O group acts as a moderate hydrogen donor. Both the uncorrected and corrected binding energies indicate that the stability of the dimers is in the order III<IV<II. This order is consistent with the proposed order based on the mean intermolecular distances.
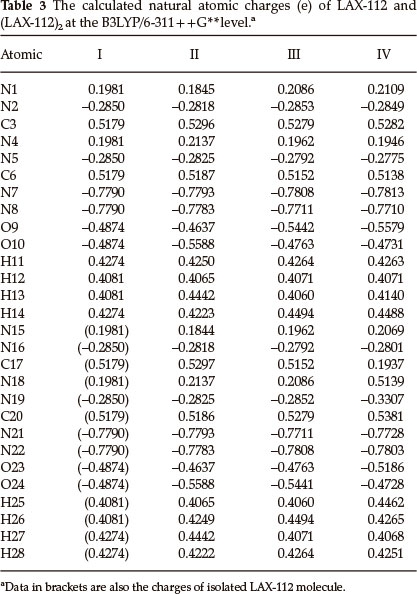
3.3 Atomic Charges and Charge Transfer
Table 3 lists the atomic NBO charges of LAX-112 and its dimers. Compared to the monomer, charges on H14 and H26 of dimer III increase by 0.0220 e, while charges on O9 and O24 decrease by 0.0567 e, indicating the effect of charge transfer by molecular contacting. There is no net charge transfer between the two sub-molecules of III due to its symmetric structure and the same distances of hydrogen bonds O9......H26 and O24......H14. Similarly, there is hardly any charge transfer between the two submolecules of the symmetrical dimer II. Charges on H14, C20 and H25 of dimer IV increase by 0.0214 e, 0.0202 e and 0.0381 e, respectively, while charges on O9, N19 and O23 decrease by 0.0705 e, 0.0457 e and 0.0312 e, respectively. The net result of charge transfer in dimer IV is that a submolecule acquires 0.0020 e. The dipole moments of LAX-112 and its dimers (II, III and IV) are 0.00, 0.00, 0.00 and 1.31 Debye. The dipole moments are zero for LAX-112, dimer II and III, due to its centrosymmetric structure.
3.4 Natural Bond Orbital Analysis
Table 4 summarizes the second-order perturbative estimates of 'donor-acceptor' (bond-antibond) interactions in the NBO basis for all the dimers. This is carried out by examining all possible interactions between 'filled' (donor) Lewis-type NBOs and 'empty' (acceptor) non-Lewis NBOs, and estimating their stabilization energy by second order perturbation theory.45-47 The stabilization energies E(2) are proportional to the NBO interaction. When a donor and an acceptor belong to different submolecules in a cluster, it is called an intermolecular NBO interaction. It is these that reveal the origin of the intermolecular interactions.

As can be seen from the intermolecular NBO interaction in Table 4, the main NBOs interacting in dimers II and III are the lone pairs on oxygen atoms of one submolecule acting as the donor and the N-H antibond of another submolecule as the acceptor. Similarly, the main NBOs interacting in dimer IV is the lone pairs on the oxygen atoms and nitrogen atom of one submolecule acting as the donor and the N-H antibond of another submolecule as the acceptor. The total stabilization energies are over 60 kJ mol-1, indicating strong hydrogen bonds. Although the stability sequence does not simply depend on the largest or the main stabilization energy, it can be concluded that the order of the stabilization energies of dimers is III>IV>II based on the total stabilization energies (II, 75.52 kJ mol-1; III, 77.08 kJ mol-1; and IV, 75.82 kJ mol-1), which is in agreement with the sequence of the hydrogen bond lengths.
3.5 Thermodynamic Properties
On the basis of vibrational analysis and statistical thermodynamics, the standard thermodynamic functions, heat capacities (cop), entropies (Som) and enthalpies (Hom), were obtained and listed in Table 5.
Both the entropy and the enthalpy change for the dimerization process are negative within the temperature range of 200.00 to 800.00 K. The intermolecular interaction is, therefore, an exothermic process accompanied by a decrease of the entropy.
The values of c°p for all the dimers are close to each other at the same temperatures, with values larger than double c°p for the monomer by 5.37~15.51 J mol-1 K-1. The values of AHT for each dimer at the same temperature give the same sequence AHTI < AHTII<AHTm as the binding energies. The change in Gibbs free energies (AGT = AHT - TAST) during the dimerization processes for the three dimers are negative under 200.0 K. Therefore, these dimers can be spontaneously produced from the isolated monomer under 200 K, and dimer II can be spontaneously produced at room temperature. The stability order of the dimers under 600 K is II>IV>III, based on the values of AGT. However, AGT gives a different stability order above 600 K, since the value of AHT is less sensitive to temperature than that of TAS, and the effect of temperature upon AGT is derived from the contributions of TAS term for the same dimer. The AGT value increases as temperature increases for each dimer, thus the interactions weaken as the temperature increases.
3.6 The Properties of Detonation Velocity and Pressure
Detonation velocity (D) and detonation pressure (P) are the most important targets in comparing the detonation characteristics of energetic materials. D and P of an explosive can be predicted with the nitrogen equivalent equation (NE equation) shown as equations (5)-(7).48
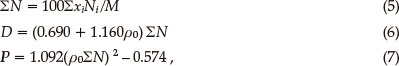
in which PN is the nitrogen equivalent of the detonation products; Nj is the nitrogen equivalent index of a certain detonation product; Xi is the moles of a certain detonation product produced by a mole of explosive; p0 is the density of an explosive (the densities of the monomer and the dimer II were obtained from the molar mass divided by the average molar volume. The average molar volumes of the compounds were obtained from the statistical average values of 100 molar volumes. The volume of each molecule, defined as the volume inside a contour of 0.001 e/bohr3 electron density,49 was calculated by the Monte Carlo method in the Gaussian 09 program package39 ).
The detonation products produced by general explosives together with their nitrogen equivalent indices are listed in Table 6. According to the order of H2-CO in forming detonation products, the detonation products of LAX-112 are calculated as follows:
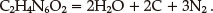
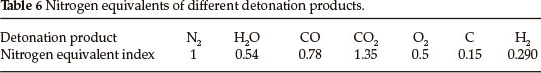
According to Eq. (5), in which M = 144.09, total nitrogen equivalents of LAX-112 are obtained through the nitrogen equivalent indices of the detonation products in Table 5:
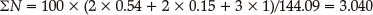
According to Eqs. (6) and (7), where ρ = 1.615 g cm-3 which was calculated based on the B3LYP/6-311 + +G** calculations, the detonation pressure (D) and detonation velocity (P) can be obtained as follows:
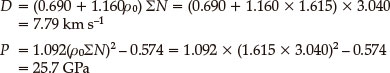
By the method above, the data of the detonation pressure and detonation velocity of II which is the most stable dimer are shown in the Table 7.

As indicated above, the calculated detonation velocity and pressure of LAX-112 monomer are 7.79 km s-1 and 25.7 GPa respectively, which are very close to the data reported by literature. The detonation velocity and pressure of dimer II are 8.02 km s-1 and 27.9 GP which are a little higher than those of the monomer. Therefore, the formation of the dimer increases the density and detonation properties of the compound.
4. Conclusions
From the DFT calculations reported above, the main conclusions can be obtained as follows:
(1) Both the binding energies and the mean intermolecular distances indicate that the stability of the dimers is in the order III<IV<II.
(2) The average binding energy of each hydrogen bond in the most stable dimer (Dimer II) is -21.19 kJ mol-1, which is larger than that of the water dimer, indicating that the hydrogen bonding is strong.
(3) Dimerization of LAX-112 is an exothermic process along with the decrease of entropies, the difference of the free energy between the monomer and dimers decrease as the temperature decreases. The process of forming the most stable dimer (dimer II) from the monomer is spontaneous at room temperature.
(4) The formation of the dimer can increase the density and detonation properties of a compound.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (21073141), the education Committee Foundation of Shaanxi Province (No 11JK0564 and 11JK0582) and Program for New Century Excellent Talents in University of Ministry of Education of China (NCET-12-1047). The authors also give much thanks to Prof. Xia Qiying (School of Chemistry and Chemical Engineering, Linyi University, Shandong, P.R. China) for the help of calculation work by G09.
References
1 S.L. Xu,S. Q. Yang,W. Zhang and X.G.Zhang, Chin. J.National University of Defense Technology, 2006, 28, 17-23. [ Links ]
2 G. Steinhauser and T.M. Klapötbe, Angew. Chem. Int. Edn., 2008, 47, 3330-3347. [ Links ]
3 E.C. David andW. Damon, Chin. J. Heterocyclic Chem, 2009,46, 88-90. [ Links ]
4 M.A. Hiskey, D.E. Chavez and D.L. Naud, US Patent 6458227, 2002. [ Links ]
5 G. Hervé, Propellants. Explos. Pyrotech., 2009, 34, 444-51. [ Links ]
6 A.S. Shawali and N.M. Tawfik, Arch. Pharm. Res., 2009, 32, 975-982. [ Links ]
7 Y.H. Gong, F. Miomandre, R. Meallet-Renault, S. Badré, L. Galmiche, J. Tang, P. Audebert and G. Clavier, Eur. J. Org. Chem., 2009, 35, 6121-6128. [ Links ]
8 G. Steinhauser and T.M. Klapötke, J. Chem. Educ., 2010, 87, 150-156. [ Links ]
9 H.X. Ma, H.M. Xiao, J.R. Song, X.H. Ju, W. Zhu and K.B. Yu, Chem. Phys., 2008, 344, 79-89. [ Links ]
10 G.Y. Fang, L.N. Xu, X.G. Hu and X. H. Li, J. Hazard. Mater, 2008, 160, 51-55. [ Links ]
11 A.N. Chermahini, A. Ghaedi, A. Teimouri, F. Momenbeik and H.A. Dabbagh, J. Mol. Struc.: THEOCHEM, 2008, 867, 78-84. [ Links ]
12 X.H. Ju, H.M. Xiao and J.Z. Tan. Chin. J. Chem., 2002, 20, 629-637. [ Links ]
13 H.M. Xiao, J. S. Li and H.S. Dong, J. Phys. Org. Chem, 2001, 14, 644-649. [ Links ]
14 H.M. Xiao, J.S. Li and H.S. Dong, Chin. Acta. Chim. Sinica, 2000, 58, 297-302. [ Links ]
15 G.Y. Fang, L.N. Xu, H.M. Xiao and X.H. Ju, Chin. Acta. Chim. Sinica, 2005, 63, 1055-1061. [ Links ]
16 L.N. Xu, H. M. Xiao, G.Y. Fang and X. H. Ju, Chin. Acta. Chim. Sinica, 2005, 63, 1062-1068. [ Links ]
17 X.H. Ju, H. M. Xiao and Q.Y. Xia, J. Chem. Phys., 2003, 119, 10247-10255. [ Links ]
18 J.S. Li, F. Zhao, F.Q. Jing and H.M. Xiao, J. Mol. Struc.: THEOCHEM,2001, 574, 213-220. [ Links ]
19 Y. Hu, H.X.Ma, J.F. Li, R. Gao and J.R. Song, Bull. Korean Chem. Soc., 2010, 31, 2897-2902. [ Links ]
20 Y. Hu, H.X. Ma, J.Q. Zhan and J.R. Song, Chin. Huaxuetongbao, 2010, 73, 263-268. [ Links ]
21 M.A. Hiskey and D.E. Chavez, Proc. 27th International Pyrotechnics Seminar: 3, July 16-21, Colorado, USA, 2000, 3-14. [ Links ]
22 L. Hans-Heinrich andR. Helmut, Energetic Mater., 1994, 12, 223-235. [ Links ]
23 M.A. Hiskey and D.E. Chavez, DE 2001776133, 2001. [ Links ]
24 D.E. Chavez andM.A. Hiskey,Heterocyclic Chem, 1998, 35, 1329-1332. [ Links ]
25 M.D.Coburn, G.A. Buntain, B.W. Harris, M.A. Hiskey, K.Y. Lee and D.G. Ott, J. Heterocyclic Chem., 1991, 28, 2049-2050. [ Links ]
26 J. Pan, J.X. He and Y.J. Tao, Chin. Energetic Mater, 2004 (Suppl.), 58-59. [ Links ]
27 S.Q. Yang and S.L. Xu, Chin. Energetic Mater., 2005, 13, 362-364. [ Links ]
28 S.L. Xu, Y.P. Lei, S.Q. Yang and W. Zhang, Chin. Energetic Mater, 2006, 14, 340-342. [ Links ]
29 M.A. Hiskey, M. Coburn and G. Donald, US Patent 5281706, 1994. [ Links ]
30 M.D. Coburn, M.A. Hiskey, K.Y. Lee, D.G. Ott and M.M. Stinecipher, J. Heterocycl. Chem., 1993, 30, 1593-1595. [ Links ]
31 Y. Zhou, X.P. Long, X. Wang, Y.J. Shu and A.M. Tian, Chin. J. Energetic Mater., 2006, 14, 315-320. [ Links ]
32 H.S. Dong and F.F. Zhou, Performance of High Energetic Explosive and Related Compounds, Science Press, Beijing, 1989. [ Links ]
33 S.L. Xu, S.Q.Yang and Y.P. Wang, Chin. J. Chemical Propellants & Polymeric Mater., 2007, 5, 14-19. [ Links ]
34 D.E. Chavez and M.A. Hiskey, J. Energetic Mater. 1999, 17, 357-377. [ Links ]
35 J. Baker, J. Comput. Chem., 1987, 8, 563-574. [ Links ]
36 T.L. Hill, An Introduction to Statistical Thermodynamics, Addison-Wesley Publishing Company, New York, 1960. [ Links ]
37 M.J. Frisch, G. W. Trucks, H.B. Schlegel, P.M.W. Gill, B.G..Johnson, M.A. Robb, J.R.Cheeseman, T. Keith, G.A. Petersson, J.A.Montgomery, K. Raghavachari, M.A.Al-Laham, VG. Zakrzewski, J.V. Ortiz, J.B. Foresman, J. Cioslowski, B.B. Stefanov, A. Nanayakkara, M. Challacombe, C.Y. Peng, P.Y. Ayala, W. Chen, M.W. Wong, J.L. Andres, E.S. Replogle, R.Gomperts, R.L. Martin, J.P. Stewart, M.H.Gordon, C. Gonzalez and J.A. Pople, Gaussisn 98, Revision A.(Gaussian, Inc, Pittsburgh ,PA, 1998). [ Links ]
38 Y. Zhao and D.G. Truhlar, Theor Chem Account. 2008, 120, 215-241. [ Links ]
39 M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, Ö. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009. [ Links ]
40 S.F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 533-566. [ Links ]
41 A. Johnson, P. Kollman and S. Rothenberg, Thermochim. Acta, 1973,29, 167-172. [ Links ]
42 G. Chalasinski and M.M. Szczesniak, Mol. Phys. 1988,63, 205-224. [ Links ]
43 H.M. Xiao and X.H. Ju, Science Press, Beijing, 2003. [ Links ]
44 M.W. Feyereisen, D. Feller and D.A. Dixon, J. Phys. Chem., 1996, 100, 2993-2997. [ Links ]
45 A.E. Reed, R.B. Weinstock and F.J. Weinhold, Chem. Phys., 1985, 83, 735-746. [ Links ]
46 A.E. Reed and F.J. Weinhold, Chem. Phys., 1985, 83, 1736-1740. [ Links ]
47 A.E. Reed, L.A. Curtiss and F.J. Weinhold, Chem. Rev. (Washington DC), 1988, 88, 899-926. [ Links ]
48 Y. Guo and H. Zhang, Explos. Shock Waves, 1983, 3, 5623-5629. [ Links ]
49 M.W. Wong, K.B. Wiberg and M.J. Frisch, Comput. Chem, 1995, 16, 385-394. [ Links ]
Received 28 January 2013
Accepted 19 May 2013
* To whom correspondence should be addressed. E-mail:donghuhai@qq.com


{kind=link}
{kind=link}