










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
RESEARCH ARTICLE
The synthesis and characterization of several corroles
Caitlin F. Zipp; Joseph P. Michael; Manuel A. Fernandes; Helder M. Marques*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, P.O. WITS, Johannesburg, 2050, South Africa
ABSTRACT
Preliminary results towards the synthesis of a corrole-based vitamin B12 analogue are reported. The synthesis of three simple corroles, 5,10,15-triphenylcorrole (TPCrl), 5,10,15-tri(2-nitrophenyl)corrole and 10-(4-methoxyphenyl)-5,15-diphenylcorrole is described. The synthesis of 10-[2-(benzoylamino)phenyl]-5,15-diphenylcorrole (DPAPCrl) suggests that a large, bulky meso substituent can be incorporated into the corrole with no loss of stability or significant decrease in yield. Both TPCrl and DPAPCrl were crystallized and their crystal structure is reported.
Keywords: Corrole, vitamin B12, X-ray diffraction crystal structure.
1. Introduction
Corroles are fully aromatic tetrapyrrole macrocycles containing a direct pyrrole-pyrrole bridge. They are the link between porphyrins, with their fully aromatic π electron system, and the corrins of vitamin B12 and its derivatives, which have a 14 π e-system delocalized over 13 atoms (Fig. 1). The absence of one of the methylene bridges of the porphyrin system gives corroles a macrocyclic cavity that is similar to that of the corrins;1 however, with an aromatic 18 π electron system, they are more electron-rich.2
Corroles are stronger acids than their porphyrin and corrin counterparts because of the presence of three pyrrole-like and one imine-like nitrogen donors; on coordination of metal ions, corroles form tri-anions, porphyrins di-anions and corrins mono-anions. They are therefore capable of binding metal ions in both very high, and very low oxidation states.2,3 The aromatic nature of corroles means the corrole ring is predominantly planar, unlike that of the porphyrins which is subject to a variety of distortions,4 or that of corrins with tetrahedral carbon atoms at the C1 and C19 positions of the ring.
Although known since1964,5 it was the independent report by two groups of a facile 'one-pot' synthesis6,7 in 1999 that led to a dramatic increase in the number of reports on corroles appearing. In spite of the differences between the two methodologies, and other methodologies that arose subsequently, the molecular basis of one-pot A3 corrole synthesis (A is the substituent at the 5, 10 and 15 positions, Fig. 1) remains the same (Scheme 1). It entails two independent steps. The first involves the reaction between pyrrole and benzaldehyde derivatives. This acid-catalyzed electrophilic substitution reaction results in a mixture of aldehyde-pyrrole polymers of various lengths with alternating pyrrole and aldehyde subunits. The second step involves the oxidative ring closure of the polymers (pyrrole-pyrrole coupling) and subsequent aromatization to yield a variety of macrocycles based on the length and identity of the polymer chain produced in the first step.8 The desired polymer for corrole synthesis is composed of the condensation of four pyrrole and three aldehyde subunits (a bilane or tetrapyrrane).8 The various one-pot synthetic methodologies may vary in the first step, but the second step is similar and entails the use of an oxidant such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) or p-chloranil (tetrachloro-1,4-benzoquinone) in a solvent such as chloroform or dichloromethane. Corrole yields are usually quite low (5-15 %).6,7,9,10

Gross and co-workers7 used a solvent-free approach in the presence of a solid support (florisil, alumina or silica, subsequently reported not be necessary for small quantities,10 but required for gram-scale syntheses). Equimolar quantities of pyrrole and aldehyde were heated for several hours; the black tar-like product was washed with dry dichloromethane and oxidized with DDQ in the presence of air. The reaction is successful provided the aldehyde contains an electron-withdrawing substituent (such as fluorine) which increases the activating inductive effect on the aldehyde, making it more reactive towards pyrrole. Yields are typically between 8 % and 11 %. The use of short bursts of microwave radiation followed by DDQ oxidation is reported in some cases to improve yields to between 30 and 80%. 11
Koszarna and Gryko reported an efficient synthetic method for the synthesis of meso-substituted corroles using a water: methanol solvent system in the presence of 0.1 equivalents of catalytic HCl.8 The method exploits a difference in solubility of the starting materials and the bilane product; whereas the starting materials and the dipyrromethane intermediate (two pyrrole units linked by a methine bridge see below) are soluble, the bilane tends to precipitate, effectively halting the reaction at the bilane stage. The bilane can then be isolated and oxidized to the corrole macrocycle. This reduces the formation of porphy-rins and other products. The use of p-chloranil rather than DDQ tended to increase corrole yields; yields of between 13 % and 56 % were reported.
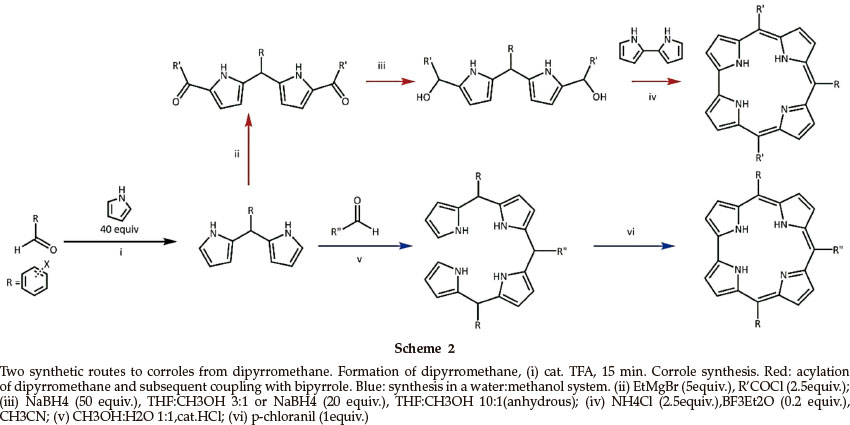
An alternative approach to corrolesis the step-wise synthesis of components of the macrocycle. Such an approach usually begins with the synthesis of a dipyrromethane from pyrrole and an aldehyde in the presence of a catalytic amount of acid, often trifluoroacetic acid (TFA);12 dipyrromethanes have been identified as important intermediates in corrole synthesis.8,13,14 The synthesis requires a large excess of pyrrole (typically 40:1-pyrrole:aldehyde), short reaction times, and low temperature (typically 15 minutes, RT) to minimize the formation of oligo- mers or pyrrole polymerization. Yields are usually better than 50 %. Dipyrromethane units can then be condensed with an aldehyde under acidic conditions to form a bilane; the presence of a chemical oxidant yields a corrole (Scheme 2),8,12,13 although porphyrins are usually formed as well. Conditions have been described under which corrole formation is favoured over that of porphyrin.13
Alternatively, the corrole macrocycle is constructed from the acylation and subsequent reduction of dipyrromethane to form a dicarbinol which is then coupled with 2,2'-bipyrrole in a [2+2] condensation to yield a corrole,14,15 with decreased chance of porphyrin formation. Yields of corrole are typically up to 12 %. The re are several routes to the synthesis of 2,2'-bipyrrole from pyrrole.16,17
Metallation can be effected after the macrocycle has been synthesized by refluxing in an appropriate solvent (for example, methanol or pyridine) with a metal acetate, chloride or carbonyl.18
We have recently started a programme to compare and contrast the chemistry of the corroles and corrins in order to elucidate the effect of the macrocyclic ring on the chemistry of Co(III). In this preliminary communication we report on the synthesis of several novel corroles which we have obtained in our exploration on the way to the synthesis of a biomimetic for aquacobalamin (vitamin B12a), which we will report on elsewhere.
2. Results and Discussion
We compared the synthesis of a simple triaryl corrole, 5,10,15-triphenylcorrole (TPCrl), using the 'one-pot' synthesis method described by Paolesse,6,9 a modification of the Lindsey method19 which is a standard method for the synthesis of porphyrins, and the solvent-free approach of Gross.7,10
In the Paolesse approach (20 mmol scale) benzaldehyde and pyrrole (3 mole equivalents) were dissolved in acetic acid and heated under reflux for three hours. The black precipitate produced was purified by column chromatography. Although NMR spectroscopic analysis indicated the presence of both 5,10,15,20-tetraphenylporphyrin (TPP) and TPCrl, we were unable to separate the very small amount of corrole from the many other unidentified products produced in spite of using a wide variety of solvents for the chromatography.
The Lindsey method19 for porphyrin synthesis was modified by altering the ratio of pyrrole:benzaldehyde from 1:1 in the typical Lindsey approach to 3:1 in an effort to favour corrole over porphyrin formation. The pyrrole and benzaldehyde were added to chloroform in the presence of catalytic BF3-Et2O under an argon atmosphere. After one hour, DDQ was added and the reaction mixture was stirred for a further hour. TPP was obtained in 8 % yield and there was no evidence (NMR, TLC) for formation of TPCrl. Two polymorphs of TPP were obtained and their structures determined by X-ray diffraction methods. There are five structural polymorphs of TTP in the Cambridge Structural Database.20,24 The two polymorphs were identical to those reported by Silvers and Tulinsky24 and by Hoard and co-workers, respec-tively.23
Not unexpectedly, the solvent-free approach of Gross7 and co-workers was unsuccessful as the aldehyde used was not derivatized with an electron-withdrawing functionality. The reaction was performed on a 2.5-mmol scale and after rapid mixing of the pyrrole and aldehyde and subsequent oxidation in dichloromethane, no corrole was detected in the resulting intractable tar.
As none of the methods appeared viable for the reliable synthesis of corroles in a reasonable yield, we turned our attention to Gryko's water:methanol solvent system in the presence of a catalytic amount of HCl.8 Pyrrole and benzaldehyde (2:1 molar ratio) were dissolved in a 1:1 methanol:water mixture in the presence of concentrated HCl (1 % volume) and stirred at room temperature in the absence of light. Subsequent extraction of the bilane into chloroform and oxidation using p-chloranil yielded after purification TPCrl in 19% yield. A small amount of TPP was also obtained.
The corrole was identified by its UV-vis spectrum25 (Fig. S1 of the Supplementary Material) with the Soret band at 412 nm and a shoulder at 432 nm, and the four bands in the visible region, characteristic of metal-free corroles and porphyrins, at 515,572, 609 and 648 nm. The parent ion m/z = ([M+H]+) was observed at at m/z = 526.98 in the LRMS. The NMR spectra of a corrole and the analogous porphyrin are readily distinguished as the four-fold symmetry of the porphyrin results in a much simpler NMR spectrum with only three distinct signals for the protons in the β positions at 8.92 ppm; the protons in the ortho positions of the meso phenyl substituents at 8.29 ppm and a complex multiplet representing the remaining meta and para protons in the 7.86-7.75 ppm region (see above). By contrast, the corrole spectrum is more complex with four separate doublets for the β protons at 8.97, 8.88, 8.60 and 8.56 ppm and two doublets and a multiplet from the meso substituents at 8.37, 8.17 and 7.857.68 ppm. The three pyrrolic protons present in the inner core of the macrocycle were observed up-field at -0.49 ppm in CDCl3.
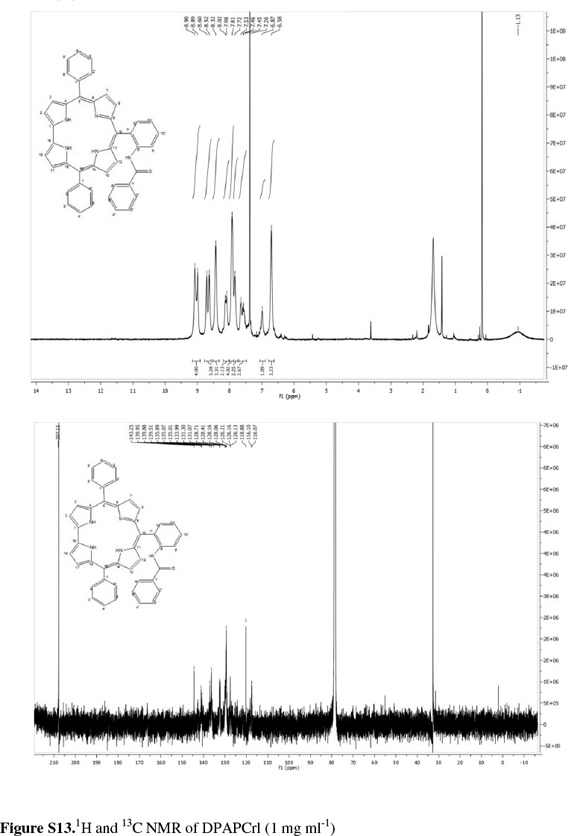
Coupling of NMR signals was only evident at very low concentrations of corrole (ca. 1mgmL-1); at higher concentrations, broad signals with no multiplet resolution were observed. We believe this arose from π...π aggregation in solution (Fig. S6 of the Supplementary Material).
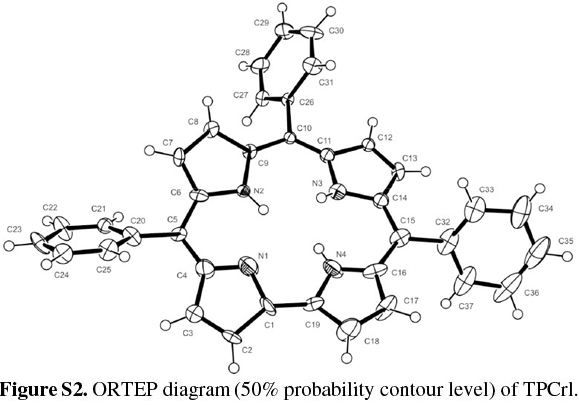
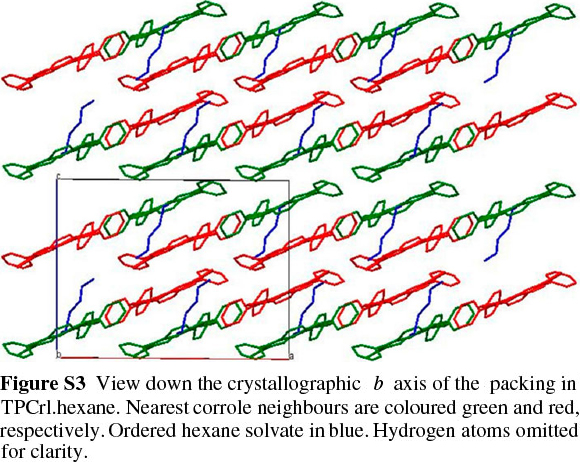
TPCrl was dissolved in dichloromethane and hexane (1:1 v/v), which was allowed to evaporate slowly at 4 °C; two different crystal structures were obtained. The first, of TPCrl itself, was crystallographically identical to that previously reported;26 the second exists as a TPCrl hexane solvate. The TPCrl hexane solvate is very similar to a previously-reported structure containing methanol, crystallizing in a similar unit cell but in the space group C2 instead of C2/c.27 The ORTEP diagram is shown in Fig. S2 of the Supplementary Material. The molecules pack in a layered structure (Fig. S3) with nearest corrole neighbours approximately 3.54 Å apart, a centroid... centroid distance of 4.34 Å and a centroid...centroid lateral displacement of 2.51 Å (Fig. 2). Table S1 of the Supplementary Material gives the bond lengths and bond angles in the molecule. As discussed in Experimental below, the presence of ordered, disordered and possibly partially absent hexane solvent made a structure solution quite difficult. Fig. S4 of the Supplementary Material shows the hex-ane solvent channels of the structure.

In the solvent-free crystal structure of TPCrl, the corroles also pack parallel to nearest neighbours, but the distance between the mean planes of the corrole N atoms is a little larger (3.88 Å) as is the centroid...centroid lateral displacement of 4.13 Å. The very close packing of the corroles is made possible by the phenyl rings rotating from perpendicular to the corrole ring, with a mean Cβ-Cmeso -C'1-C2' torsion angle of 50(3) °. (There is much greater variation in the solvent-free structure - 50(15) ° - with one phenyl substituent at 64.9 ° significantly closer to the perpendicular because the packing of the corrole molecules is not as close.) This close packing of molecules observed in the solid state, and our observation that there is very significant line broadening of the NMR signals at concentrations >1 mg mL-1, suggests that π...π interactions are significant in simple metal-free corroles. In order to accommodate the three H atoms in the macrocyclic cavity, the corrole ring is distorted from planar with the Cβ atoms of pyrrole rings alternatively above and below the mean plane of the corrole. This type of distortion is also observed in porphy-rins, and is referred to as a saddled (sad) distortion.4
TPCrl was metallated with cobalt(II) acetate in the presence of triphenylphosphine to give a five-coordinate complex with one triphenylphosphine ligand in 51 % yield, confirmed by single crystal X-ray diffraction, identical to the structure published by Paolesse and co-workers in 1999.6 The parent ion was observed in the LRMS at m/z = 844.82 ([M+H]+) and the characteristic change in UV-vis spectrum upon metal complexation was observed (Fig. S1 of the Supplementary Material). The presence of the large triphenylphosphine ligand ensured that the macro-cycles did not aggregate in solution or in the solid state. As a result, the 1H NMR spectrum consisted of well-resolved multiplets. The sharp, well-defined signals gave clear evidence of the diamagnetic nature of the cobalt ion in the 3+ oxidation state.
We explored the scope of Gryko's water:methanol method by synthesizing a corrole with a strongly electron-withdrawing group in the phenyl substituents, 5,10,15-tri(2-nitrophenyl)corrole, TNPCrl. The yield was only 4.5 %. We were unable to crystallize the corrole and had to resort to spectroscopic evidence to confirm its identity. The 13C NMR spectrum showed resonances at 152.37 and 152.25 pm, consistent with C2'(NO2); The NO stretch was observed at 1541 cm-1; and the LRMS spectrum showed m/z = 662([M+H]+).TNPCrlhaspreviouslybeenreportedin8-10 % yield28 using a modified version of Paolesse's original synthesis.9
The one-pot synthetic methods available were of limited utility for our purposes. In our experience, yields tended to be low and purification was often problematic. The method is also limited in its scope in that the product obtained would have three identical substituents at the meso positions (Scheme 1). We therefore turned our attention to the step-wise approach of Scheme 2 which proceeds though a dipyrromethane intermediate, and in particular the route involving a bilane intermediate.
The synthesis of a dipyrromethane can produce trimers, tetramers and higher oligomers. We found that the formation of the longer chain species was limited, but not avoided, by using a large excess of pyrrole (40 mole equivalents) as well as a short reaction time (15 min).12 The reaction of benzaldehyde and excess pyrrole produced a brown solid that was isolated in 57 % yield and was identified as 2,2-(phenylmethylene)bis(1H-pyrrole), a simple aryl dipyrromethane. There were no aldehyde signals from benzaldehyde in the NMR spectrum; the methine proton was observed as a singlet at 5.43 ppm in the 1H NMR spectrum and at 43.97 ppm in the 13C NMR spectrum. The positions of the pyrrole signals were shifted up-field shift with three signals observed at 6.65, 6.15 and 5.90 ppm, instead of the two signals of unsubstituted pyrrole at 6.74 and 6.24 ppm.
To verify the synthesis of an A2B corrole (A = substituent at C5 and C15, B = substituent at C10) we used Gryko's methanol: water synthesis strategy to prepare 10-(4-methoxyphenyl)-5,15-diphenyl-corrole (DPMPCrl) in 29 % yield. We were unable to crystallize the corrole and identified it from its LRMS with m/z = 557.06 ([M+H]+). There is also a characteristic singlet at4.09 ppm due to the methoxy group in the 1H NMR spectrum and a band at 1247 cm-1 due to the C-O stretch in the infrared spectrum.
The development of a corrole model for vitamin B12a requires the incorporation of a tail in the corrole which terminates in an aromatic N-donor ligand capable of coordinating Co(III). To test the feasibility of this, we set out to design and synthesize a corrole with a bulky substituent at the 10 position.
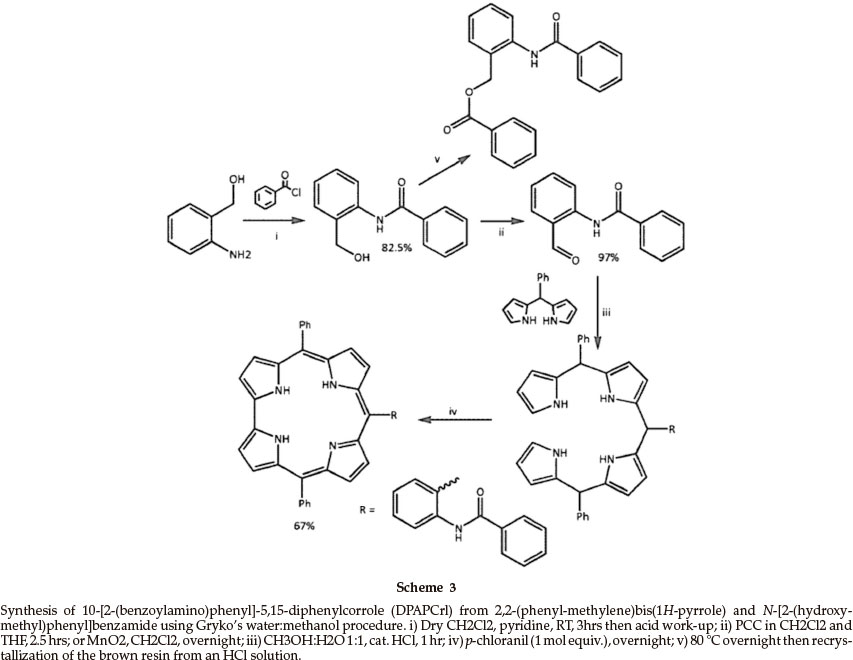
N-[2-(Hydroxymethyl)phenyl]benzamide was synthesized from 2-aminobenzyl alcohol (the structure of which we have published elsewhere29) and benzoyl chloride in 82 % yield, based on the work of Fürstner and Jumbam (Scheme 3).30 The amide was identified from the down-field NH signal at 9.82 ppm in the 1H NMR spectrum and the carbonyl signal at 165.76 ppm in the 13C spectrum. The parent ion was observed at m/z = 227.0944 ([M]+) in the high-resolution mass spectrum. Subjecting the alcohol to high temperatures for an extended period of time (80 °C, overnight) resulted in the formation of a brown resin with the crystallization of benzoic acid. Recrystallization of the resin from an aqueous solution of hydrochloric acid yielded colourless needle-like crystals. The product, 2-(benzamido) benzylben-zoate,was identified using 1H NMR; the hydroxyl proton which was present at 4.07 ppm in the 1H NMR spectrum of N-[2-(hydroxymethyl)phenyl]benzamide was absent. In addition, the methylene proton signal shifted from 4.66 to 5.40 ppm because ofthepresence of the more electron rich carboxylic ester. The signals in the aromatic region integrated for 14 aromatic protons, indicating the incorporation of a third phenyl ring.
Oxidation of N-[2-(hydroxymethyl)phenyl]benzamide to the aldehyde, N-(2-formylphenyl) benzamide, was achieved in 97 % yield using manganese dioxide in chloroform over a 12-hour period. The presence of the aldehyde was confirmed by the down-field aldehyde proton signal at 10.03 ppm in the 1H NMR spectrum as well as the carbonyl peak at 195.92 ppm in the 13C NMR spectrum, typical for aldehyde signals. The parent ion was observed at m/z= 225.0786 ([M]+) in the high-resolution mass spectrum.
The corrole10-[2-(benzoylamino)phenyl]-5,15-diphenyl-corrole (DPAPCrl) was synthesizedfrom N-(2-formylphenyl) benzamide and dipyrromethane2,2-(phenylmethylene) bis(1H-pyrrole) using the methanol:water method in 67 % yield. The NMR spectrum, while complex, was typical for a corrole macrocycle with signals from the β protons at 8.98, 8.89, 8.60 and 8.52 ppm and signals from the aromatic substituent maintained. The parent ion was present at m/z = 646.42 ([M+H]+)inthe LRMS.
N-[2-(Hydroxymethyl)phenyl]benzamide, N-(2-formyl-phenyl)benzamide and 2-(benzamido)benzyl benzoate were all found to be crystalline and polymorphic. The polymorphism resulted from differences in the van der Waals associations in the solid state, as discussed elsewhere.31
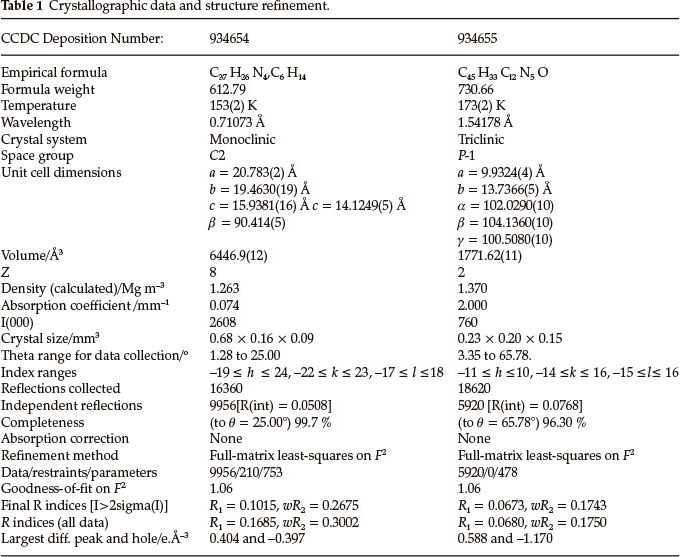
DPAPCrl crystallized from dichloromethane; the asymmetric unit contains one DCM solvent molecule (Fig. 3). The unit cell is shown in Fig. S5 of the Supplementary Material. The crystallo-graphic details are listed in Table 1. The bond lengths and angles are given in Table S2 of the Supplementary Material.
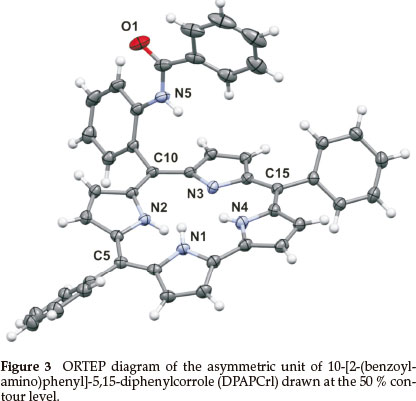
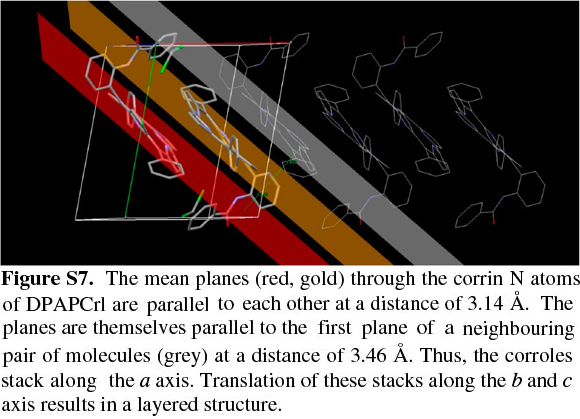
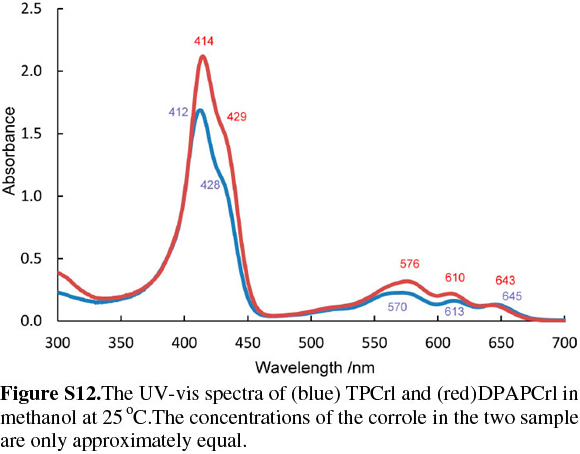
The corrole itself, as observed with TPCrl (see above) is sad distorted, presumably to accommodate the three NH protons in the macrocyclic cavity. Two corrole molecules are packed in a face-to-face manner about a centre of inversion in the unit cell (Fig. S5 of the Supplementary Material). The mean planes through the four N atoms of each corrole are parallel to each other, separated by 3.14 Å. These planes are themselves parallel to the first plane of a neighbouring pair of molecules, at a distance of 3.46 Å (Fig. S7). Thus, the corroles stack along the a axis. Translation of these stacks along the b and c axis results in a layered structure. We observed the same concentration-dependent line-broadening effects in the NMR as with TPCrl, suggesting that this aggregation persist in solution (Fig. S6 of the Supplementary Material). The UV-vis spectrum of DPAPCrl is very similar to that of TPCrl, suggesting that the pendant tail amide is not conjugated to the corrole (Fig. S12 of the Supplementary Material) and the solution structure is therefore probably similar to that shown in the solid state.
3. Summary and Conclusions
The feasibility of synthesizing a corrole-based vitamin B12 analogue has been established, firstly with the synthesis of simple corroles, including 5,10,15-triphenylcorrole, 5,10,15-tri(2-nitrophenyl)corrole and 10-(4-methoxyphenyl)-5,15-diphenyl-corrole. The synthesis of 10-[2-(benzoylamino)phenyl]-5,15-diphenylcorrole suggests that a large, bulky meso substituent can be incorporated into the corrole with no loss of stability or significant decrease in yield.
The corroles prepared were synthesized using Gryko's metha-nol:water procedure.8 This was found to be far superior to the one-pot methods proposed in 1999 and the early 2000s.6,9,11,13 This versatile method can be used in the preparation of both A3 and trans A2B corroles.
The significant π..π overlap of the macrocycles in both the solid state and in solution was brought to light by the discovery of the concentration-dependence of the signal resolution in the NMR spectra. This was confirmed by the close intermolecular packing of the corroles in the solid state in crystal structures of 5,10,15-triphenylcorrole and 10-[2-(benzoylamino)phenyl]-5,15-diphenylcorrole.
The synthesis of a vitamin B12 corrole analogue will be reported elsewhere.
4. Experimental
Unless otherwise indicated, all syntheses were conducted in a dark room under dim red light conditions.
4.1. Synthesis of 5,10,15-Triphenylcorrole (TPCrl)
4.1.1. The Paolesse Approach6
Freshly distilled pyrrole (4.2 mL, 60.5 mmol) and benz-aldehyde (2.0 mL, 20 mmol) were added to glacial acetic acid (250 mL). The stirred colourless solution was refluxed for 3 hours and became dark green-black. After cooling to room temperature deionized water (250 mL) was added and a precipitate was filtered off. Solid NaCl was added to the filtrate to produce a further precipitate. The combined precipitates were separated by silica gel flash chromatography with dichloromethane as eluent. TPP, TPCrl,2,2-(phenylmethylene)bis (1 H-pyrrole) were identified by 1HNMR analysis among other unidentified products. A number of attempted chromatographic separations using different eluents (ethyl acetate:hexane (1:4 v/v) containing 1 % triethylamine; ethyl acetate:hexane (1:4 v/v); ethyl acetate: petroleum ether (1:4 v/v) containing 1 % triethylamine; ethyl acetate; methanol) were unsuccessful in separating the low yield of corrole from the other products.
4.1.2. Modified Lindsey Method
Freshly distilled pyrrole (0.208 mL, 3.0 mmol) and benz-aldehyde (0.102 ml, 1.0 mmol) were added to dry chloroform (400 mL); this was deaerated (Ar) for 15 min and BF3(OEt2) (168 µl) was added. An immediate colour change from colourless to yellow-green was observed. The flask was covered in foil and the reactuion mixture stirred at room temperature for 1 hour by which time the solution had turned orange. DDQ (173 mg, 0.76 mmol) was washed into the flask with chloroform (150 mL). The colour of the solution immediately changed to black. The reaction mixture was stirred for an hour at room temperature, then concentrated in vacuo. The resulting mixture was separated by column chromatography on flash silica gel with ethyl acetate:hexane (1:9 v/v) containing 1 % triethylamine as eluent to yield tetraphenylporphyrin as a purple-black solid (14.1 mg, 8 %). Melting point >250 °C. For standard numbering of porphyrins see Fig. S8 of the Supplementary Material. δH 300 MHz, CDCl3): 8.92 (8H, s, pyrrole Hβ), 8.29 (8H, dd, J=7.1, 1.8 Hz, Harortho), 7.86-7.75 (12H, m, Harmeta and para). δC (75 MHz, CDCl3; assignments are tentative): 142.21 (C1'), 134.62 (C2' and C6'), 131.24(C), 127.76 (C4'), 126.74 (C3' & C5'), 120.20 (C5, C10, C15 and C20). LRMS (ESI) m/z : 615.25([M+H]+), calc.: 615.25. vmax (cm-1): 3314 (w, NH stretch); 980(s, NH bend); 964 (porphyrin skeleton ring vibration); 794(s, C-H out of plane bend); 694 (vs, NHbend in plane). UV-vis (nm, in DCM), 414; 430 (sh); 519; 571; 616; 650.
4.1.3. The Solvent-free Method of Gross7
A solution of freshly distilled pyrrole (0.173 mL, 2.5 mmol) and benzaldehyde (0.254 mL, 2.5 mmol) in dichloromethane (2 mL) was vigorously stirred at room temperature for 1 min. The tar-like mixture was dissolved in dichloromethane (50 mL) and oxidized with DDQ (0.283 g, 1.25 mmol) for one hour. There was no evidence of the formation of a macrocycle in the 1H NMR spectrum.
4.1.4. Gryko's Synthesis in 1:1 Water:Methanol8
Freshly distilled pyrrole (0.700 mL, 10 mmol) and benzaldehyde (0.510 mL, 5 mmol) were dissolved in methanol (200 mL). Hydrochloric acid (32 %, 4.25 mL) in water (200 mL) was added to the reaction flask which was stoppered and kept in the dark. The mixture was stirred at ambient temperature for three hours after which time an orange precipitate had formed. The reaction mixture was extracted into chloroform. The red organic layer was washed with water twice, dried with Na2SO4 and diluted with chloroform to a volume of 600 mL. p-Chloranil (1.23 g, 5 mmol) was added and the reaction mixture was refluxed for 1 hour in the dark. The black reaction mixture was allowed to cool to room temperature and concentrated in vacuo. The crude product mixture was separated by column chromatography on flash silica gel with dichloromethane as eluent. The corrole-containing fractions (identified using UV-vis spectroscopy) were combined and evaporated to dryness in vacuo. A dark purple-black powder was obtained (83.2 mg; 19 %). Melting point > 250 °C. For standard numbering of corroles see Fig S9 of the Supplementary Material. δH (500 MHz, CDCl3): 8.97 (2H, d br, pyrrole Hβ), 8.88 (2H d br, pyrrole Hβ), 8.60 (2H, d br, pyrrole Hβ), 8.56 (d 2H, d br, pyrrole Hβ), 8.37 (4H, d, J=5.6 Hz, Har), 8.17(2H, d br, Har), 7.85-7.68 (9H, m,Har), -0.49 (3H, s br, NH). δC(126 MHz, CDCl3): 143.13, 142.14, 140.86, 139.82, 139.29, 135.96, 135.03, 131.55, 128.05, 127.40, 125.97, 122.65, 118.72, 115.81, 109.73. LRMS (APCI) m/z: 526.98 ([M + H]+) 524.91 ([M-H]-); calc.: 527.22 ([M+H]+). vmax (cm-1): 2942 (s, NH stretch); 2844 (s, CH stretch); 1716 (w, C=N stretch); 1454 (m, C=C stretch); 1344 (m, CH bend), 1047 and 1013 (CN stretch and aromatic ring stretches and bends).
4.2. Preparation of Triphenylphosphine [5,10,15-Triphenylcorrolato]cobalt(III), [CoTPCrl(PPh3)]6
5,10,15-Triphenylcorrole (25 mg, 0.0475 mmol), cobalt(II) acetate (25 mg, 0.1 mmol) and triphenylphosphine (25 mg, 0.095 mmol) were stirred in methanol under reflux conditions (25 mL) for 1 hour in the dark. The reaction was monitored using UV-vis spectroscopy. The solvent was evaporated in vacuo and the crude product was separated using column chromatography on flash silica gel with dichloromethane as eluent. The red fraction was collected and concentrated in vacuo. The solid was recovered (0.0205 g, 51.1 %) and recrystallized from a hexane: dichloromethane (1:1 v/v) solvent system as deep red block-like crystals. Meltingpoint > 250 °C.δH (500 MHz, CDCl3): 8.59 (2H, d, J=4.3 Hz, pyrrole Hβ), 8.32 (2H, d, J=4.7Hz, pyrrole Hβ), 8.09 (4H, d, J=4.7Hz, pyrrole Hβ), 8.02 (2H, d, J=3.9 Hz, Har), 7.99 (1H, d, J=7.2 Hz, Har), 7.69-7.47 (m, 11H, Har), 7.37 (1H, d, J=7.3 Hz, Har), 7.04 (3H, t, J=6.8 Hz, para-Har PPh3), 6.70 (6H, dd, J=7.3, 6.0 Hz, meta-Har PPh3), 4.72 (6H, dd, J=9.7, 8.5 Hz, ortho-Har PPh3). δC(126 MHz, CDCl3): 146.27; 145.59; 145.56; 145.01; 143.41; 141.88 (Ca + meso) 136.44, 131.77 (Car PPh3), 131.70 (Car PPh3), 131.53 (Cβ pyrrole), 130.89 (Car), 130.73 (Car), 129.84, 129.29 (Car PPh3), 129.27 (Car PPh3), 128.54; 127.61; 127.35; 127.12; 127.08; 126.99; 126.96 (Car &Car PPh3) 125.38; 124.73; 124.37; 124.28; 124.25; 124.10; 123.04; 123.00; 118.57 (Cβ pyrrole). LRMS(APCI) m/z: 844.79 ([M + H]+); calc.: 845.22 ([M + H]+). νmax (cm-1): 2947 (s, CH stretch); 1715 (w, C=N stretch); 1427 (m, C=C stretch); 1311 (m, CH bend), 1050 and 1015 (CN stretch and aromatic ring stretches and bends). Single crystal XRD (identical to CSD refcode KIMQOM): Space group P-1; a = 8.84 Å, b = 13.12 A c 18.20Å; a = 94.33 °, β = 92.29 °, γ = 97.72o; R1 = 3.22 %.
4.3. Synthesis of 5,10,15-Tri(2-nitrophenyl)corrole (TNPCrl)
The synthesis of TNPCrl was by Gryko's water:methanol method. Freshly distilled pyrrole (0.697 mL, 10 mmol) and 2-nitrobenzaldehyde (0.756 g, 5 mmol) were dissolved in methanol (200 mL). Hydrochloric acid (32 %, 4.25 mL) in water (200 mL) was added to the reaction flask which was stoppered and kept in the dark. The reaction mixture was stirred at ambient temperature for three hours after which time an orange precipitate had formed. The reaction mixture was extracted into chloroform. The red organic layer was washed with water twice, dried with Na2SO4and diluted with chloroform to a volume of 600 mL. ρ-Chloranil (1.23 g, 5 mmol) was added and thereaction was refluxed for one hour in the dark. The black reaction mixture was allowed to cool to room temperature and concentrated in vacuo. The crude product mixture was separated by column chroma- tography on flash silica gel with dichloromethane as eluent. The corrole-containing fractions (identified using UV-vis spectros-copy) were combined and evaporated to dryness in vacuo and a green-black solid was obtained (0.0745 g, 4.5 %). Melting point >250 °C. δH (500 MHz, CDCl3): 8.91 (2H, dd, J=10.2, 3.9 Hz, pyrrole Hβ), 8.64-8.54 (2H, m, pyrrole Hβ), 8.33 (8H, dt, J=25.0, 7.9 Hz, pyrrole Hβ and Har), 8.27-8.22 (2H, m, Har), 7.98-7.81 (6H, m, Har), -1.98 (3H, s, NH). δC (126 MHz, CDCl3): 152.37, 152.25, 137.11, 137.06, 136.02, 133.71, 131.76, 131.67, 131.59, 131.50, 131.41, 131.19, 131.00, 129.21, 129.14, 126.76, 125.85, 125.74, 124.60, 124.54, 1 24.45, 124.03, 123.90, 116.35, 105.65, 105.52. LRMS (APCI) m/z: 662 ([M+H]+); calc.: 662 ([M+H]+). vmax (cm-1): 2981 (s, NH stretch); 2906 (s, CH stretch); 1701 (w, C=N stretch); 1541 (m, N=O stretch); 1456 (m, C=C stretch); 1344 (m, CH bend), 1062 and 1045 (CN stretch and aromatic ring stretches andbends). UV-vis (nm, in DCM): 413; 427 (sh); 576; 614; 650.
4.4. Routes to Corroles via a Dipyrromethane and a Bilane
4.4.1. Preparation of 2,2-(Phenylmethylene)bis(1H-pyrrole) (dipyrromethane)12
Benzaldehyde (0.2 mL, 2 mmol) was dissolved in a 40 mol excess of freshly distilled pyrrole (5.6 mL, 80 mmol) and was degassed (Ar) for 10 min. Trifluoroacetic acid (16 µ1 mmol) was added. The solution was stirred at room temperature for 15 min and then diluted with dichloromethane (100 mL) and washed with 0.1 M NaOH solution (100 mL). The organic layer was dried with Na2SO4 and concentrated in vacuo to remove the excess pyrrole. The brown oil was separated by column chroma-tography on flash silica gel with hexane:ethyl acetate (4:1 v/v) containing 1 % triethylamine as eluent. The desired product (0.252 g, 57 %) was obtained as a light brown solid. P98-100 °C (reported 98°C32). δH (300 MHz, CDCl3):7.84 (2H, br s, NH), 7.36-7.12 (5H, m, Har), 6.65 (2H, H5, dd, J=4.1, 2.6), 6.15 (2H, H4, dd, J=5.9, 2.8), 5.90 (2H, H3, br s,), 5.43 (1H, s, PhCH). δC (75 MHz, CDCl3): 13C(75 MHz, CDCl3δ (ppm): 142.10 (C1'), 132.54 (C2), 128.68 (C2' and C6'), 128.44 (C3' and C5'), 127.02 (C4'), 117.28 (C5), 108.44(C4), 107.27(C3), 43.97(PhCH). LRMS (ESI): 23.10 ([M + H]+); calc.: 223.12.HRMS(EI) m/z: 222.1150; calc.: 222.1157.vmax (cm-): 3363 (s, N-H stretch), 3339 (s, Ar C-H stretch), 2962 (w, C-H stretch), 1554 (m, Ar ring stretch), 1398 (w, C-H bend), 886 (s, Ar C-H bend), 777 (s, Ar C-H bend).
4.4.2. Preparation of 10-(4-Methoxyphenyl)-5,15-diphenylcorrole
2,2'-(Phenylmethylene)bis(1H-pyrrole) (0.222 g, 1 mmol) and 4-methoxybenzaldehyde (0.061 mL, 0.5 mmol) were dissolved in methanol (50 mL). Hydrochloric acid (32 %, 2.5 mL) in water (50 mL) was added to the reaction flask which was stoppered and kept in the dark. The reaction mixture was stirred at ambient temperature for 1 hour and then extracted into chloroform. The organic layer was washed with water twice, dried with Na2SO4 and diluted with chloroform to a volume of 250 mL. p-Chloranil (0.369 g, 1.5 mmol) was added and the reaction mixture was stirred at ambient temperature overnight in the dark. The black reaction mixture was concentrated in vacuo. The crude product mixture was separated by column chromatography on flash silica gel with dichloromethane as eluent. The corrole-containing fractions (identified using UV-vis spectroscopy) were combined and evaporated to dryness in vacuo to give a black solid product (0.0794 g, 29 %). MP > 250 °C. δH (500 MHz, CDCl3): 8.97 (2H, d broad, pyrrole Hβ), 8.88 (2H, d broad, pyrrole Hβ), 8.58 (4H, d broad, pyrrole Hβ), 8.37 (4H, d broad, Har), 8.09 (2H, d broad, Har), 7.81 (5H, s, Har), 7.72 (3H, s, Har), 4.09 (3H, s, CH3). δC (126 MHz, CDCl3): 140.75, 136.57, 131.84, 130.82, 130.76, 129.60, 129.25,128.92, 128.45, 128.31, 128.04, 127.87, 127.57, 127.39, 126.03, 117.57, 115.66, 113.99,112.81, 55.35 (CH3).LRMS(APCI) m/z: 557.06 ([M+H]+); calc.: 557.23 ([M+H]+). vmax (cm-1): 2976 (s, NH stretch); 2871 (s, CH stretch); 1683 (w,C=Nstretch); 1510 (m, C=C stretch); 1350 (m, CH bend), 1247 (C-O stretch); 1058 and 1018 (CN stretch and aromatic ring stretches and bends).UV-vis (nm, in DCM): 417; 434(sh); 521; 572; 621; 654.
4.5. Synthesis of 10-[2-(Benzoylamino)phenyl]-
5,15-diphenylcorrole (DPAPCrl).
4.5.1. Preparation of N-[2-(hydroxymethyl)phenyl]benzamide30 2-Aminobenzyl alcohol (5 mmol, 0.615 g) and benzoyl chloride (6 mmol, 0.635 mL) were dissolved in dry dichloromethane (15 mL) in the presence of pyridine (6.25 mmol, 0.552 mL). The reaction mixture was stirred at room temperature for three hours. HCl (0.1 N, 10 mL) was added. The aqueous layer was extracted into dichloromethane, washed with aqueous saturated NaHCO3 solution (20 mL) and water (10 mL), and dried with Na2SO4. The off-white solid crystallized on evaporation of solvent in vacuo (0.937 g, 82.5 %). MP94-95 °C (reported 93-94 °C33). See Fig. S10. δH (300 MHz, CDCl3): 9.82 (1H, s, NH), 8.14 (1H, d, J=8.1 Hz, H6), 7.84 (2H, d, J=7.3 Hz, H2' and H6'), 7.47 (2H, d, J=7.4 Hz, H3' and H5'), 7.38 (1H, t, J=7.5 Hz, Har), 7.30-7.20 (1H, m, Har), 7.06 (1H, dd, J=7.3, 1.2 Hz, Har), 6.99 (1H, dd, J=10.7, 4.0 Hz, Har), 4.66 (2H, s, CH2), 4.07 (1H, s, OH). δC-(75 MHz, CDCl3): 165.76 (C=O), 137.78 (C2), 134.51 (C1'), 131.89 (C1), 129.67 (Car), 128.94 (Car), 128.76 (C2' & C6'), 128.62 (Car), 127.13 (C3' & C5'), 124.25 (Car), 122.13 (Car), 64.60 (CH2). LRMS(ESI) m/z: 228.08 ([M+H]+); calc.: 228.10. HRMS(EI) m/z: 227.0944 ([M]+); calc.: 227.0946. vmax (cm-1): 3292 (m, br, O-H and N-H stretch (H-bonded)); 3054 (aromatic stretch); 1656 (C=O stretch); 1588 (N-H bend).
4.5.2. Preparation of N-(2-formylphenyl)benzamide N-[2-(Hydroxymethyl)phenyl]benzamide (455 mg, 2 mmol) and manganese(IV) oxide (2.608 g, 30 mmol) were stirred in chloroform (10 mL) for 12 hours. The reaction mixture was filtered through celite with chloroform as eluant. The solvent was removed in vacuo. The oily residue crystallized when placed under high vacuum. The off-white solid was recrystallized from methanol and hexane (0.436 g, 96.9 %). MP67-69 °C and 71-72 °C (reported 67-68 °C34). Melting points are of the two different polymorphs.31 See Fig. S11. δΗ (300 MHz, CDCl3): 12.11 (1H, s, NH), 10.03 (1H, s, CHO), 8.97 (1H, d, J=8.4 Hz,H3), 8.19-7.97 (3H, m, Η3', H4'& H5'), 7.78-7.65 (2H, m, Har), 7.57 (2H, dd, J=10.5, 4.5Hz, HJ, 7.32-7.24 (1H,m, HJ. δC (75 MHz, CDCl3): 195.92 (CHO), 166.17 (C=O amide), 141.28 (C2), 136.42 (C1), 136.22 (C1'), 134.34 (Car), 132.23 (Car), 128.90 (Car), 127.53 (Car), 123.07 (Car),122.01 (Car), 119.99 (CarLRMS (APCI) m/z:): 226.04 ([M+H]+); calc.: 226.08 ([M+H]+). HRMS(EI) m/z: 25.0786 ([M]+); calc.: 225.0790. vmax(cm-1): 3318 (w, N-H stretch); 1741 (m, C=O); 1526 (s, N-H stretch).
4.5.3. Preparation of 10-[2-(Benzoylamino)phenyl]-5,15-diphenylcorrole (DPAPCrl).
2,2-(Phenylmethylene)bis(1-Hpyrrole) (0.053 g, 0.24 mmol) and N-[2-formylphenyl]benzamide (0.027 g, 0.12 mmol) were dissolved in methanol (15 mL). Hydrochloric acid (32 %, 4.25 mL) in water (15 mL) was added to the reaction flask which was stoppered and kept in the dark. The reaction mixture was stirred at ambient temperature for 1 hour after which time a red precipitate had formed. The reaction mixture was extracted into chloroform. The amber organic layer was washed with water twice, dried with Na2SO4and diluted with chloroform to a volume of 60 mL. p-Chloranil (0.08 g, 0.33 mmol) was added and the reaction was stirred at ambient temperature in the dark overnight. The crude product mixture was concentrated in vacuo and separated by column chromatography using flash silica gel with dichloromethane as eluent. The corrole-containing fractions (identified using UV-vis spectroscopy) were combined and evaporated to dryness in vacuo to yield a green-black solid (0.0525 g, 67 %). MP> 250 °C. δΗ (500 MHz, CDCl3): 8.98 (2H, s*, pyrrole Ηβ), 8.89 (2H, s*, pyrrole Ηβ), 8.60 (2H, s*, pyrrole Ηβ), 8.52 (2H, s*, pyrrole Ηβ), 8.32 (1H + 2H, s*, NH + Har), 8.00 (2H, d*, J=18.6 Hz, Har), 7.81 (5H, s*, Har), 7.72 (2H,s*, Har), 7.58-7.41 (m, 3H, Har), 6.87 (1H, s*, Har tal), 6.58 (4H, s*, Har tal), -1.13 (s, 3H, 3 x NH). Signals marked * presented as very broad peaks with poor resolution. These signals are expected to be multiplets. δC ((126 MHz, CDCl3): 207.13(C=O), 143.25, 139.95, 139.88, 139.51, 135.89, 135.07, 135.01, 131.30, 131.07, 128.71, 128.41, 128.16, 128.06, 126.21, 126.16, 126.13, 118.88, 116.10, 116.07. LRMS(APCI) m/z: 646.42 ([M+H]+); calc.: 646.26. vmax(cm-1): 2981, 2964, 2947 (m, N-H stretch); 2873, 2877 (m, aromatic stretch); 1668 (w, C=O); 1058, 1031 (N-C stretch). UV-vis (nm, in DCM): 416; 431 (sh); 522; 580; 613; 649.
4.6. Materials and Methods
All reagents were purchased from Merck, Sigma-Aldrich or Aldrich and were of the highest purity available. They were used as received unless otherwise indicated. Benzaldehyde was dried over magnesium sulfate overnight, filtered, and distilled under N2 at reduced pressure and stored under N2. Pyrrole was dried over calcium hydride overnight, then distilled using fractional distillation using a Vigreux column under N2 or Ar; the purified liquid was stored under N2 or Ar, covered in foil at -18 °C. When dry solvents were required, acetonitrile, dichloromethane and dimethylformimide were distilled from calcium hydride; diethyl ether and tetrahydrofuran were distilled from sodium wire; and toluene was distilled from sodium lumps. Deionized water was purified using a Direct Q UV 3 Millipore and further purified using a Millipore MilliQ unit. Separation of compounds by column chromatography was performed using flash silica-gel 60 (particle size 0.04-0.063 mm).
Melting points were recorded using a melting point apparatus with microscope fitted with a JMinst 628 digital thermometer. A heating rate of 2 °C min-1 was used. Melting points are reported without correction.
Proton NMR data were recorded at 300.13 MHz and carbon NMR data were recorded at 75.47MHz on a BrukerAVANCE 300 spectrometer. Spectra of corrole macrocycles were recorded on a Bruker ULTRASHIELD 500 PLUS spectrophotometer where proton NMR data were recorded at 500.13 MHz and carbon NMR data were recorded at 125.77 MHz. All samples were dissolved in CDCl3 unless otherwise indicated. Chemical shifts are reported on the δ scale in ppm relative to the internal standard 0.03 % tetramethylsilane (1H) and the central CDCl3 line at 77.00 ppm (13C). NMR spectroscopic data were processed using the MestReNova software suite.35
Infra-red spectra were recorded on a Bruker Tensor 27 infrared spectrophotometer fitted with a diamond ATR device. The acquisitionof spectra was facilitated by Bruker OPUS software.36
Low-resolution mass spectra were recorded using either an APCI or ESI ion source in both the negative and positive modes on a Finnigan LXQ linear ion trap mass spectrometer using a direct injection method. High-resolution data were collected in the positive mode using an EI ion source on a Thermo Fisher high-resolution magnetic sector DFS mass spectrophotometer with a resolution of 3000, a mass range of 100 amu (8 kV) and a scan rate of 6 sec/decade.
Single crystal X-ray diffraction intensity data intensity data for TPCrl were collected at -120(2) °C on a Bruker APEX II CCD area detector diffractometer with graphite monochromated Mo Ka radiation (50 kV, 30 mA). The collection method involved ω-scans of width 0.5 ° and 512 x 512 bit data frames. Intensity data for DPAMCrl were collected at -100(2) °C on a Bruker X8 Proteum diffractometer equipped with a PLATINUM 135 CCD detector and Motel 200 optics, using Cu Ka radiation (45 kV, 20 mA) from a Microstar rotating-anode generator. The collection method involved ω-scans of width 0.5 ° and 1024 x 1024-bit data frames. Data reduction was carried out using the program SAINT+.37 All crystal structures were solved by direct methods. Non-hydrogen atoms were first refined isotropically followed by anisotropic refinement by full matrix least-squares calculations based on F2. Hydrogen atoms for DPAPCrl were first located in the difference map then positioned geometrically and allowed to ride on their respective parent atoms in the final refinements.
The structure solution of TPCrl was not trivial and several approaches were used to find the structure best fitting the available data. The highest symmetry space group in which the structure will solve is C2/c with one full corrole molecule and one full hexane molecule emerging from the structure refinement. This solution results in a R1 value of 15 %. Applying the SQUEEZE algorithm38,39 and refining the structure leads to a R1 value of about 12.5 %. At this point we attempted a solution in the space group C2. In this case the structure solution and refinement leads to a R1 value of 11.6 % with two corrole molecules and one hexane molecule in the asymmetric unit, together with very weak electron density near the molecules. Examining the structure for void space indicated the presence of solvent channels along the c axis of the structure surrounded by the ordered hexane molecules as well as the corrole molecules (Fig. S9 of the Supplementary Material). The identification of two sites for the hexane molecule (one ordered and one disordered) indicates that the correct space group for the structure is C2. The SQUEEZE algorithm was then applied to the data which accounted for 223 electrons or about 2.6 hexane molecules in channels. The contribution of the extra electron density has been added to the crystal density, chemical formula and F000 in the final refinements. The refinement of the structure in C2 unfortunately leads to a lower data to parameter ratio with the result that some of the thermal ellipsoids are rather large. Attempts have been made to minimize these effects with the use of SHELXL, DELU and ISOR restraints with the final R1 value being 10.2 % but some A and B level alerts remain. All hydrogen atoms were positioned geometrically and allowed to ride on their respective parent atoms in this structure. Diagrams and publication material were generated using SHELXTL40, PLATON,39 ORTEP-341 and MERCURY.42
Abbreviations Used
DPAPCrl, 10-[2-(benzoylamino)phenyl]-5,15-diphenyl-corrole; TNPCrl, 5,10,15-tri(-nitrophenyl)corrole; TPCrl, 5,10,15-triphenylcorrole; TPP, 5,10,15,20-tetraphenylporphyrin.
Acknowledgements
This work as made possible by funding from the Mellon Foundation, the University of the Witwatersrand, and the DST/NRF South African Research Chairs Initiative. Dr Richard Mampa is thanked for recording the NMR spectra.
References
1 D.T. Gryko, Eur. J. Org. Chem, 2002, 1735-1743. [ Links ]
2 I. Aviv-Harel and Z. Gross, Chem. Eur. J, 2009, 15, 8382-8394. [ Links ]
3 C.A. Joseph and P.C. Ford, J. Am. Chem. Soc, 2005, 127, 6737-6743. [ Links ]
4 O.Q. Munro, J.C. Bradley, R.D. Hancock, H.M. Marques, F. Marsicano and P.W. Wade, J. Am. Chem. Soc., 1992, 114, 7218-7230. [ Links ]
5 A.W Johnson and I.T Kay, J. Chem. Soc. A, 1965, 1620-1629. [ Links ] 6. R. Paolesse, L. Jaquinod, D.J. Nurco, S. Mini, F. Sagone, T. Boschi and K.M. Smith, Chem. Commun, 1999, 1307-1308. [ Links ]
7 Z. Gross, N. Galili and I. Saltsman, Angew. Chem. Int. Edn., 1999, 38, 1427-1429. [ Links ]
8 B. Koszarna and D.T. Gryko, J. Org. Chem., 2006, 71, 3707-3717. [ Links ]
9 R. Paolesse, S. Nardis, F. Sagone and R.G. Khoury, J. Org. Chem., 2001, 66, 550-556. [ Links ]
10 Z. Gross, N. Galili, L. Simkhovich, I. Saltsman, M. Botoshansky, D. Blaser, R. Boese and I. Goldberg, Org. Lett., 1999, 1, 599-602. [ Links ]
11 J.P. Collman and R.A. Decréau, Tetrahedron Lett., 2003, 44, 1207-1210. [ Links ]
12 C.-H. Lee and J.S. Lindsey, Tetrahedron, 1994, 50, 11427-11440. [ Links ]
13 D.T. Gryko and K. Jadach, J. Org. Chem, 2001, 66, 4267-4275. [ Links ]
14 P.D. Roa, S. Dhanalekshmi, B.J. Little and J.S. Lindsey, J. Org. Chem., 2000, 65, 7323-7344. [ Links ]
15 R.A. Decréau and J.P. Collman, Tetrahedron Lett.,2003, 44, 3323-3327. [ Links ]
16 T. Itahara, J. Chem. Soc., Chem. Commun., 1980, 49-50. [ Links ]
17 L. Fu and G.W. Gribble, Tetrahedron Lett., 2008, 49, 7352-7354. [ Links ]
18 C. Erben, S. Will and K.M. Kadish, Metallocorroles: Molecular structure, spectroscopy and electronic states, in The Porphyrin Handbook, Vol. 2, (K.M. Kadish, K.M. Smith and R. Guilard, eds.), Academic Press, New York, 2000, [ Links ]
19 J.S. Lindsey, Synthesis of meso-substituted porphyrins, in The Porphyrin Handbook, vol. 1, (K.M. Kadish, K.M. Smith and R. Guilard eds.), Academic Press, New York, 2000. [ Links ]
20 S.J. Silvers and A. Tulinsky, J. Am. Chem. Soc, 1967, 89, 3331-3337. [ Links ]
21 G. Dunnay and C.B. Storm, Mol. Crystallogr, 1967, 2, 287-291. [ Links ]
22 K. Kano, K. Fukuda, H. Wakami, R. Nishiyabu and R.F. Pasternack, J. Am. Chem. Soc., 2000, 122, 7494-7502. [ Links ]
23 M.J. Hamor, T.A. Hamo and J.L. Hoard, J. Am. Chem. Soc., 1964, 86, 1938-1942. [ Links ]
24 B.L. Barker, G.G. Stanley and F.R. Fronczek, Personal communication, CCDC 188202, Refcode TPHPOR11, 2002. [ Links ]
25 C.J. Ziegler, J.R. Sabin, G.R. Geier and VN. Nemykin, Chem. Commun., 2012, 48, 4743-4745. [ Links ]
26 T. Ding, J.D. Harvey and C.J. Ziegler, J. Porphyrins Phthalocyanines, 2005, 9, 22-27. [ Links ]
27 R. Paolesse, A. Marini, S. Nardis, A. Froiio, F. Mandoj, D.J. Nurco, L. Prodi, M. Montalti and K.M. Smith, J. Porphyrins Phthalocyanines, 2003, 7, 25-36. [ Links ]
28 J.P. Collman and R.A. Decréau, Org. Lett, 2005, 7, 975-978. [ Links ]
29 C.F. Zipp, M.A. Fernandes, H.M. Marques and J.P. Michael, Acta Cryst., 2012, E68, o174. [ Links ]
30 A. Fürstner and D.N. Jumbam, Tetrahedron, 1992, 48, 5991-6010. [ Links ]
31 C. Zipp, H. Dirr, M. Fernandes, H.M. Marques and J.P. Michael, Cryst. Growth & Design, submitted. [ Links ]
32 J.S. Lindsey, L. Yu, P. Thamyongkit and A.D. Bhise, USA, Patent US2007/155963 A1, 2007. [ Links ]
33 O. Kirino, S. Yamamoto and T. Kato, Agric. Biol. Chem., 1980, 44, 2143-2147. [ Links ]
34 R.A. Kumar, C.U. Maheswari, S. Ghantasala, C. Jyothi and K.R. Reddy, Adv. Synth. Cat, 2011, 353, 401-410. [ Links ]
35 MestReNova, 6.0.2-5475, Mestrelab Research S.L., Santiago de Compostela, 2009. [ Links ]
36 OPUS, 5.5.60, Bruker Optik GmbH, Ettlingen, Germany, 2005. [ Links ]
37 SAINT + (includes XPREP and SADABS), 6.0, Bruker AXS Inc., Madison, Wisconsin, USA, 2005. [ Links ]
38 P. van der Sluis and A.L. Spek, Acta Cryst, 1990, A46, 194-201. [ Links ]
39 A.L. Spek, J. Appl. Cryst, 2003, 36, 7-13. [ Links ]
40 SHELXTL (includes XS, XL, XP, XSHELL), 5.1, Bruker AXS Inc., Madison, Wisconsin, USA, 1999. [ Links ]
41 L.J. Farrugia, J. Appl. Cryst, 1997, 30, 565-566. [ Links ]
42 C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Cryst., 2008, 41, 466-470. [ Links ]
Received 19 April 2013
Revised 23 May 2013
Accepted 28 May 2013
* To whom correspondence should be addressed. E-mail: helder.marques@wits.ac.za
Supplementary Information for The Synthesis and Characterisation of Several Corroles
S. Afr. J. Chem., 2013, 66, 158-166
Caitlin F. Zipp, Joseph P. Michael, Manuel A. Fernandes and Helder M. Marques*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, P.O. Wits, Johannesburg, 2050 South Africa












{kind=link}
{kind=link}
{kind=link}
{kind=link}