










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban ago. 2013
RESEARCH ARTICLE
Synthesis and biological evaluation of novel thiosemicarbazone-triazole hybrid compounds as antimalarial agents
Henok H. Kinfe*; Yonas H. Belay
Department of Chemistry, University of Johannesburg, P.O. Box 524, Auckland Park, 2006 South Africa
ABSTRACT
A novel series of thiosemicarbazone-triazole hybrids were efficiently synthesized and evaluated for their activity against the 3D7 strain of the malaria parasite, Plasmodium falciparum. Although the hybrids were found not to be as potent as the standard chloroquine, they have shown activities interesting enough to warrant future structure activity relationship (SAR) studies.
Keywords: Thiosemicarbazone, triazole, hybrid, antimalarial.
1. Introduction
Malaria is an infectious disease claiming the lives of millions of people in the developing countries. According to the recent World Health Organization (WHO) report there are between 300 million and 500 million cases of malaria worldwide annually and more than 1 million people die from the disease, most of them children under the age of five years.1 Ninety per cent of the cases and 75 % of the deaths occur in sub-Saharan Africa.1 Chloroquine, a 4-aminoquinoline, has been the most reliable and effective drug for the treatment of malaria for much of the past five decades. Even though the drug has several advantages including limited host toxicity, low cost and effective and well-established synthesis protocols, its use has been hampered by the emergence of chloroquine-resistant strains of malaria.2,3 Currently, artemisinin-based combination therapy (ACT) is WHO's standard treatment against Plasmodium falciparum malaria, the most lethal species that cause malaria in humans, in which the regimen uses a double or triple combination therapy with the aim of either delaying or preventing the development of drug resistance.4-6 Although no clinical resistance has been registered against artemisinins, recent reports from South-East Asia are increasingly pointing to tolerance, which may eventually lead to resistance against this class of drugs.7 The widespread emergence of drug-resistant strains calls for the search for new drugs preferably with novel mechanisms of action.8 One approach in tackling some of the problems has been via the use of the so-called hybrid drugs, which comprise two or more drug pharmacophores in one molecule with the intention to exert multi-drug action.9 For instance, trioxaquines are synthetic hybrid molecules containing two covalently linked pharmacophores (1,2,4-trioxane and an aminoquinoline), a concept referred to as 'covalent biotherapy', and thus possesses a dual mode of action, namely heme alkylation with the trioxane entity, and heme stacking and inhibition of haemozoin formation with the aminoquinoline moiety.10,11 The trioxaquines exhibited improved antimalarial activity over their individual fragments, indicating a potentially synergistic effect of the hybrids.10 In the pursuit of novel hybrid drugs against malaria, we are interested in the synthesis of thiosemicarbazone and triazole hybrid compounds which to the best of our knowledge have not been reported in the literature. Thiosemicarbazones, having several metal binding sites, possess a wide spectrum of biological activity which includes antiviral,12 anticancer13 and antimalarial14 activities probably mediated via chelation to intracellular cations. Equally important are triazoles which can interact with biological targets through hydrogen bonding and dipole interactions.15 They are the main structural features of a number of antifungal drugs (e.g. fluconazole, isavuconazole, itraconazole, variconazole, pramiconazole, ravuconazole and posaconazole).16 Besides their potent antifungal activities, they also exhibit analgesic, anti-inflammatory, local anaesthetic, anticonvulsant, antineoplastic, antiviral and antimalarial activities.17-19
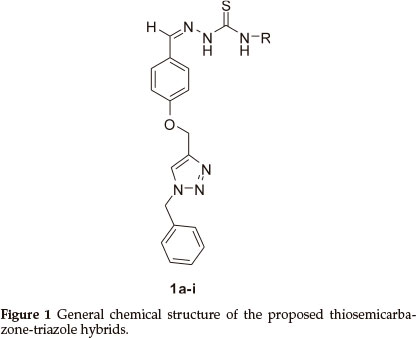
These clinically important activities of thiosemicarbazone and triazole moieties prompted us to investigate the antimalarial activity of the hybrid structure of the two. Herein, we report the synthesis of such novel hybrids 1a-i (Fig. 1) having various R groups and their activity against P. falciparum.
2. Results and Discussion
2.1 Synthesis
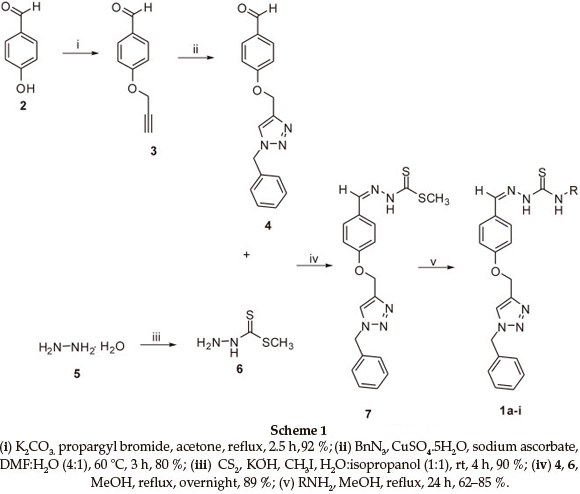
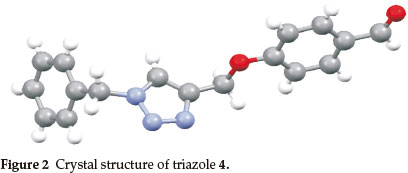
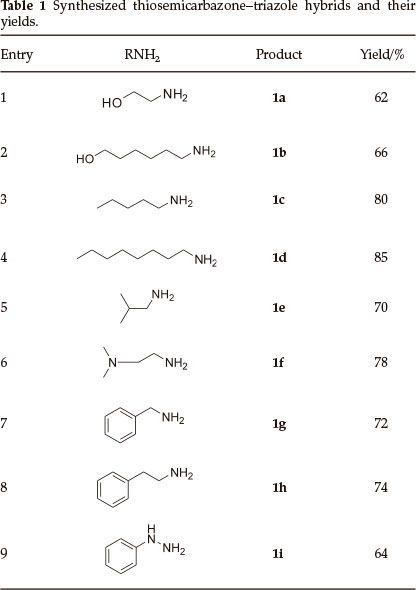
The synthesis of the target hybrid structure is outlined in Scheme 1. The synthesis commenced with alkylation of commercially available 4-hydroxybenzaldehyde with propargyl bromide in the presence of K2CO3 to give 3 which incorporates an alkynyl group required for click chemistry.20 Compound 3 was then subjected to click chemistry with freshly prepared benzyl azide to provide 1,4-disubstituted triazole 4.21 The structure of the triazole was established using 1H NMR spectroscopy. Disappearance of the signal for the terminal alkyne proton at 2.54 ppm and appearance of the characteristic signal for the alkene proton of the triazole ring at 7.53 ppm were evidence in support of the structure. X-ray crystallographic analysis further confirmed that the triazole formed was the 1,4-disubstituted isomer as shown in Fig. 2 (see experimental section for deposition details). Methylhydrazinecarbodithioate 6, which was prepared in a one pot synthesis from the condensation of hydrazine monohydrate, CS2 and methyl iodide,22 was reacted with triazole 4 under Schiff's base condensation reaction conditions to give compound 7.22 The structure of the compound was established using NMR spectroscopy. Disappearance of the signal due to the alde-hydic proton at 9.85 ppm and carbonyl carbon signal at 190.8 ppm from triazole 4, and appearance of a methyl signal at 2.50 ppm and C=S signal at 197.6 ppm in the 1H and 13C NMR spectra, respectively, confirmed formation of compound 7. The latter then underwent nucleophilic substitution reactions with a series of primary amines to provide a library of hybrid compounds 1a-i as shown in Table 1.
2.2. In vitro Antiplasmodial Activity
The synthesized hybrid compounds were evaluated for their in vitro antiplasmodial activity against 3D7 strain of the malaria parasite, P. falciparum, using chloroquine as a reference drug. The percentage parasite viability results after exposure of the P. falciparum to the synthesized hybrid compounds is summarized in Fig. 3 and Table 2. Unfortunately, out of all the compounds tested, only compound 1f exhibited less than 50 % parasite viability (46 % compared to 38 % of chloroquine). The remaining compounds displayed more than 50 % parasite viability with 1a showing no activity at all. Compounds with longer aliphatic chains such as 1b, 1c and 1d exhibited lower percentage parasite viability (56, 61 and 61 %, respectively) when compared to compounds with shorter alkyl chains such as 1a and 1e which exhibited 68 and 100 %, respectively. Similar results were also obtained with the aryl amine groups where extension of the carbon chain in 1g by one to 1h resulted in almost 20 % decrease in parasite viability.
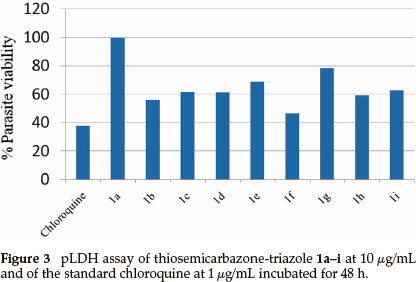
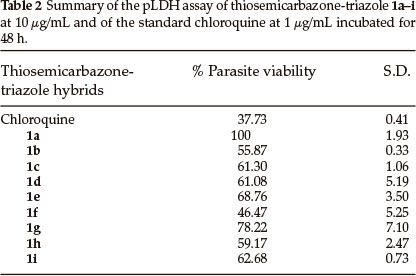
3. Conclusion
Novel thiosemicarbazone-triazole hybrids were successfully synthesized and evaluated for their antiplasmodial activity. While the compounds turned out not to be very potent, they showed interesting activity, thus warranting further investigation. We are currently trying to improve activity by synthesising analogues of 1f, which is the most active hybrid in the series and our results will be reported in due course.
4. Experimental
All the solvents used were AR grade and all the reagents were purchased from Sigma Aldrich. The reactions were monitored by thin layer chromatography (TLC) on aluminium-backed Merck silica gel 60 F254 plates using an ascending technique when required. The plates were visualized under UV light. Gravity column chromatography was done on Merck silica gel 60 (70-230 mesh). Melting points were determined using a Reichert-Jung Thermovar hot-stage microscope and are uncor-rected. Infrared spectra were recorded using Tensor 27 Brucker and Perkin Elmer FT-IR spectrum BX. Elemental analyses were performed on a Vario Elementar III microcube CHNS analyzer at Rhodes University, South Africa. All proton nuclear magnetic resonance (1H NMR) spectra were recorded as CDCl3 or DMSO-d6 solutions (unless indicated otherwise) using tetramethyl silane as an internal standard on a Bruker Ultrashield (400 MHz) spectrometer. Carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded on the same instruments at 100 MHz using tetramethylsilane as an internal standard. All chemical shifts are reported in ppm.
Methylhydrazinecarbodithioate (6)
To an ice-cooled solution of KOH (22.41 g, 399.3 mmol) in 50 mL of H2O:isopropanol (1:1) was added hydrazine monohydrate (19.40 mL, 399.2 mmol) followed by a drop-wise addition of CS2 (24.10 mL, 399.9 mmol) while maintaining the temperature between 0-10 °C. After stirring for three hours, ice-cooled CH3I (24.90 mL, 400.0 mmol) was added drop-wise and the reaction mixture was left stirring for four hours. The resulting precipitate was filtered, washed several times with cold water and recrystallized from dichloromethane to provide 6 as a white crystalline solid in 90 % yield; mp 80-83 °C (Lit. 81-83 °C)22; 1H NMR (CD3OD, 400 MHz): δ 2.52 (s, 3H, CH3); 13C NMR (CD3OD, 100 MHz): δ 194.5 (C=S), 17.2 (CH3). The spectroscopic data were in agreement with the reported data.23
4-Prop-2-ynyloxybenzaldehyde (3)
A solution of 4-hydroxybenzaldehyde 1 (2.53 g, 20.5 mmol) in acetone (20 mL) was treated with K2CO3 (4.21 g, 30.8 mmol). The resulting reaction mixture was stirred under reflux for 30 min followed by the addition of propargyl bromide (5.20 mL, 45.1 mmol of 80 wt.% in toluene). After stirring for 2 hours, the reaction mixture was concentrated in vacuo. The residue was then taken up in ethyl acetate, washed successively with water and brine, and dried over Na2SO4. The crude product was purified by column chromatography (ethyl acetate/hexane, 2:8) to give the title compound as a white solid in 92 % yield; mp 75-78 °C (Lit. 79-80 °C)24; 1H NMR (CDCl3, 400 MHz): δ 9.88 (s, 1H, CHO), 7.83 (d, 2H, J = 8.0 Hz, Ar), 7.07 (d, 2H, J = 8.0 Hz, Ar), 4.76 (s, 2H, CH2), 2.54 (s, 1H, C=CH); 13C NMR (CDCl3, 100 MHz): δ 190.7 (C=O), 162.3, 131.8, 130.5, 115.1 (Ar), 77.5 (C=CH), 76.3 (C=CH), 55.9 (CH2OAr). The spectroscopic data were in agreement with the reported data.20
1-[4-(1-Benzyl-1 H-[1,2,3]triazol-4-ylmethoxy)-phenyl]-ethanone (4)
To a solution of 4-prop-2-ynyloxybenzaldehyde 3 (2.02 g, 12.5 mmol) in DMF:water (35 mL, 4:1) freshly prepared benzyl azide (4.70 mL, 37.5 mmol), CuSO4.5H2O (0.15 mmol, 37 mg, dissolved in 0.50 mL of water) and sodium ascorbate (1.0 mmol, 0.2 g dissolved in 1 mL of water) were successively added at 60 °C and the resulting reaction mixture was stirred at the same tem- perature for 3 h. After completion of the reaction, the reaction mixture was transferred into a separatory funnel containing brine (30 mL) and the aqueous layer was extracted with ethyl acetate (3 x 40 mL). The combined organic layers were dried over anhydrous Na2SO4 and the resulting crude product was purified by column chromatography (ethyl acetate/hexane, 4:6) to give the title compound in 80 % yield as white crystals; mp 100-103 °C; 1H NMR (CDCl3,400 MHz): δ 9.85 (s, 1H, CHO), 7.79 (d, 2H, J=8.4 Hz, Ar), 7.54 (s, 1H, H-triazole), 7.35-7.24 (m, 5H, Ar), 7.05 (d, 2H, J=8.4 Hz, Ar), 5.51 (s, 2H, CH2OAr), 5.23 (s, 2H, ArCH2N); 13C NMR (CDCl3,100 MHz): δ 190.8 (C=O), 163.1, 134.6, 132.0, 130.3, 129.2, 128.9, 128.1, 115.0 (Ar), 143.6 (N-C=CHN), 122.8 (N-C=CHN), 62.1 (ArCH2N), 54.3 (CH2OAr). The spectroscopic data were in agreement with the reported data.25
(Z)-methyl-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl) methoxy]benzylidene} hydrazine carbodithioate (7)
A solution of compound 4 (2.1 g, 6.8 mmol) in methanol (20 mL) was treated with methylhydrazine carbodithioate 6 (0.81 g, 6.8 mmol). The reaction mixture was stirred overnight under reflux. The hot solution was then poured into an ice-water mixture and the precipitate formed was filtered and successively washed with water and dichloromethane to provide the title compound in 89 % yield as a yellow solid; mp 204-205 °C; IR (neat, cm-1): 3130, 1600, 1304, 1220, 1059and 825; 1H NMR (DMSO-d6, 400 MHz): δ 13.20 (bs, 1H, NH), 8.28 (s, 1H, H-triazole), 8.17 (s, 1H, ArCH=N), 7.64 (d, 2H, J = 8.4 Hz, Ar), 7.37-7.29 (m, 5H, Ar), 7.10 (d, 2H, J = 8.4 Hz, Ar), 5.59 (s, 2H, CH2OAr), 5.18 (s, 2H, ArCH2N), 2.5 (s, 3H, CH3); 13C NMR (DMSO-d6, 100 MHz): δ 197.6 (C=S), 160.2, 136.0, 129.2, 128.9, 128.3, 128.0, 126.3, 115.4 (Ar), 146.3 (N-C = CHN), 142.7 (ArCH=N), 124.9 (N-C=CHN), 61.3 (ArCH2N), 53.0 (CH2OAr), 16.8 (CH3); Anal. Calcd. for C19H19N5OS2: C, 57.41; H, 4.82; N, 17.62; S, 16.13 %. Found: C, 54.38; H, 4.98; N, 16.97; S, 15.26 %.
General Procedure for the Synthesis of the Hybrid Compounds 1a-i
A solution of compound 7 (0.50 g, 1.3 mmol) in methanol (10 mL) was treated with the alkyl amines (13.0 mmol) and the resulting reaction mixture was stirred overnight under reflux. The hot solution was then poured into an ice-water mixture and the precipitate formed was filtered and recrystallized from a mixture of dichloromethane and hexane to afford hybrids 1a-i:
(Z)-2-{4-[(1-benzyl-1 H-1,2,3-triazol-4-yl)methoxy]benzylidene}-N-(2-hydroxyethyl)hydrazinecarbothioamide (1a): 62 % yield; white solid; mp 206-208 °C; IR (neat, cm-1): 3355,3151,1605,1280,1224, 1049 and 827; 1H NMR (DMSO-d6, 400 MHz): δ 11.41 (bs, 1H, NH), 8.36 (bs, 1H, CH2NH), 8.30 (s, 1H, H-triazole), 7.99 (s, 1H, ArCH=N), 7.71 (d, 2H, J = 8.4 Hz, Ar), 7.37-7.30 (m, 5H, Ar), 7.07 (d, 2H, J = 8.0 Hz, Ar), 5.6 (s, 2H, CH2OAr),5.17 (s, 2H, ArCH2N), 4.81 (bs, 1H, OH), 3.61 (d, 2H, J = 5.2 Hz, HNCH2CH2OH), 3.55 (d, 2H, J = 4.8 Hz, HNCH2CH2OH); 13C NMR (DMSO-d6 100 MHz): δ 176.9 (C=S), 159.4, 136.0, 128.8, 128.2, 128.0, 127.0, 114.9 (Ar), 142.7(N-C=CHN), 141.9 (ArCH=N), 124.8 (N-C=CHN), 61.2 (ArCH2N), 59.3 (HNCH2CH2OH), 52.9 (CH2OAr), 45.9 (HNCH2CH2OH). Anal. Calcd. for C20H22N6O2S: C, 58.52; H, 5.40; N, 20.47; S, 7.81 %. Found C, 58.13; H, 5.76; N, 19.85; S, 8.10 %.
(Z)-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy]benzylidene}-N-(6-hydroxyhexyl)hydrazinecarbothioamide (1b): 66 % yield; white solid; mp 146-148 °C; IR (neat, cm-1): 3407,3364,3145,1604,1297, 1218, 1052 and 824; 1H NMR (DMSO-d6, 400 MHz): δ 11.24 (bs, 1H, NH), 8.45 (t, 1H, J = 5.4 Hz, CH2NH), 8.28 (s, 1H, H-triazole), 7.97 (s, 1H, ArCH=N), 7.72 (d, 2H, J = 8.4 Hz, Ar), 7.38-7.29 (m, 5H, Ar), 7.05 (d, 2H, J = 8.4 Hz, Ar), 5.59 (s, 2H, CH2OAr),5.16 (s, 2H, ArCH2N), 4.50-4.35 (m, 1H,OH), 3.37 (d, 2H, J = 5.6 Hz, HN(CH2)5CH2OH), 1.57-1.10 (m, 10H, 5 x CH2); 13C NMR (DMSO-d6, 100 MHz): δ 176.7 (C=S), 159.5, 136.1, 129.0, 128.9, 128.4, 128.1, 127.2, 115.0 (Ar), 142.9 (N-C = CHN), 141.9 (ArCH=N), 124.9 (N-C=CHN), 61.3 (ArCH2N), 60.8 (alkyl CH2), 53.0 (CH2OAr), 43.6, 32.6, 29.2, 26.4, 25.5 (5 x CH2); Anal. Calcd. for C24H30N6OS: C, 61.78; H, 6.48; N, 18.01; S, 6.87 %. Found: C, 61.21; H, 6.76; N, 17.84; S, 6.61 %.
(Z)-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy]benzylidene}-N-pentylhydrazinecarbothioamide (1c): 80 % yield; white solid; mp 147-149 °C; IR (neat, cm-1): 3303, 3170,1604,1298,1223,1050 and 824; 1H NMR (CDCl3), 400 MHz): δ 9.71 (bs, 1H, NH), 7.79 (s, 1H, ArCH=N), 7.54 (s, 1H, H-triazole), 7.52 (s, 2H, Ar), 7.39-7.34 (m, 3H, Ar), 7.25 (d, 2H,J = 7.2 Hz, Ar), 6.96 (d, 2H,J = 8.4 Hz, Ar), 5.51 (s, 2H, CH2OAr), 5.19 (s, 2H, ArCH2N), 3.80-3.60 (m, 2H, HNCH2(CH2)3CH3), 1.80-1.52 (m, 2H, HNCH2CH2(CH2)2CH3) 1.50-1.24 (m, 4H, HNCH2CH2(CH2)2CH3), 0.89 (s, 3H, CH3); 13C NMR (CDCl3, 100 MHz): δ 177.0 (C=S), 160.0, 134.3, 129.1, 128.8, 128.1, 126.5, 115.1 (Ar), 144.0 (N-C = CHN), 142.1 (ArCH=N), 122.7 (N-C=CHN), 62.1 (ArCH2N), 54.3 (CH2OAr), 44.5, 29.0, 28.9, 22.3 (alkyl CH2), 14.0 (CH3); Anal. Calcd. for C23H28N6OS: C, 63.28; H, 6.46; N, 19.25; S, 7.34 %. Found: C, 63.32; H, 6.79; N, 18.82; S, 7.05 %.
(Z)-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy]benzylidene}-N-octylhydrazinecarbothioamide (1d): 85 % yield; white solid; mp 145-147 °C; IR (neat, cm-1): 3330, 3149,1605,1296,1231,1054 and 831; 1H NMR (CDCl3, 400 MHz): δ 9.62 (bs, 1H, NH), 7.78 (s, 1H, ArCH=N), 7.55 (s, 1H, H-triazole), 7.52 (s, 2H, Ar), 7.40-7.34 (m, 3H, Ar), 7.25 (d, 2H, J = 9.2 Hz, Ar), 6.96 (d, 2H, J = 8.4 Hz, Ar), 5.51 (s, 2H, CH2OAr), 5.19 (s, 2H,ArCH2N), 3.80-3.60 (m, 2H, HNCH2(CH2)6CH3),1.75-1.56 (m, 2H, HNCH2CH2(CH2)5CH3), 1.50-1.12 (m, 10H, HNCH2CH2(CH2)5CH3), 0.92-0.3 (m, 3H, CH3); 13C NMR (CDCl3,100 MHz): δ 176.6 (C=S), 159.9, 134.1, 129.2, 128.9, 128.2, 126.5, 115.1 (Ar), 143.8 (N-C = CHN), 142.4 (ArCH=N), 122.9 (N-C=CHN), 61.9 (ArCH2N), 54.4 (CH2OAr), 53.4,44.5,31.7,29.2,29.1,26.9,22.6 (CH2), 14.1 (CH3); Anal. Calcd. for C26H34N6OS: C, 65.24; H, 7.16; N, 17.56; S, 6.70 %. Found: C, 64.89; H, 7.84; N, 17.50; S, 6.49 %.
(Z)-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy]benzylidene}-N-isobutylhydrazinecarbothioamide (1e): 70 % yield; white solid; mp 174-175 °C; IR (neat, cm-1): 3375, 3137,1605,1290,1211,1051 and 833; 1H NMR (CDCl3, 400 MHz): δ 9.82 (bs, 1H, NH), 7.81 (s, 1H, ArCH=N), 7.53 (s, 1H,H-triazole), 7.52 (s, 2H, Ar), 7.34 (s, 3H, Ar), 7.25 (d, 2H, J = 4.4 Hz, Ar), 6.96 (d, 2H, J = 8.0 Hz, Ar), 5.51 (s, 2H, CH2OAr), 5.19 (s, 2H, ArCH2N), 3.53 (s, 2H, HNCH2), 2.151.87 (m, 1H, HNCH2CH(CH3)2), 1.20-0.85 (m, 6H,2xCH3); 13C NMR (CDCl3, 100 MHz): δ 177.2 (C=S), 160.0, 134.3, 129.1, 128.8, 128.1, 126.5, 115.1 (Ar), 144.0 (N-C = CHN), 142.2 (ArCH=N), 122.7 (N-C=CHN), 62.1 (ArCHN), 54.3 (CH2OAr), 51.7 (HNCH2), 28.3 (HNCH2CH(CH3)2), 20.3 (CH3); Anal. Calcd. for C22H26N6OS: C, 62.53; H, 6.20; N, 19.89; S, 7.59 %. Found: C, 62.03; H, 6.70; N, 19.78; S, 7.27 %.
(Z)-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy]benzylidene}-N-(2-(dimethylamino)ethyl)hydrazinecarbothioamide (1f): 78 % yield; white solid; mp 179-180 °C; IR (neat, cm-1): 3360, 3165, 1604, 1275,1212,1050 and 831; 1H NMR (DMSO-dfe 400 MHz): δ 11.36 (bs, 1H, NH), 8.35 (bs, 1H, CH2NH), 8.28 (s, 1H, H-triazole), 7.98 (s, 1H, ArCH=N), 7.69 (d, 2H, J = 8.0 Hz, Ar), 7.34-7.31 (m, 5H, Ar), 7.06 (d, 2H, J = 8.0 Hz, Ar), 5.59 (s, 2H, CH2OAr),5.16 (s, 2H, ArCH2N), 3.62 (d, 2H, J = 5.6 Hz, HNCH2CH2), 2.44 (s, 2H, HNCH2CH2), 2.17 (s, 6H;2xCHs); 13C NMR (DMSO-d6, 100 MHz): δ 176.8 (C=S), 159.6, 136.1, 128.9, 128.4, 128.1, 127.2, 115.1 (Ar), 142.9 (N-C = CHN), 142.1(ArCH = N), 124.9 (N-C=CHN), 61.3 (ArCH2N), 53.0 (CH2OAr), 57.8 (HNCH2CH2), 45.4 (CH3), 41.42 (HNCH2CH2); Anal. Calcd. for C22H27N7OS: C, 60.39; H, 6.22; N, 22.41; S, 7.33 %. Found: C, 59.92; H, 6.57; N, 21.91; S, 7.12 %.
(Z)-N-benzyl-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl]methoxy}benzyl idenehydrazinecarbothioamide (1g): 72 % yield; white solid; mp 200-202 °C; IR (neat, cm-1): 3363, 3134,1604,1291,1224,1051 and 829; 1H NMR(DMSO-d6,400 MHz): δ 11.50 (bs, 1H, NH), 9.03 (t, 1H, J = 6.2 Hz, CH2NH), 8.28 (s, 1H, H-triazole), 8.03 (s, 1H, ArCH=N), 7.75 (d, 2H, J = 8.8 Hz, Ar), 7.35-7.22 (m, 10H, Ar), 7.05 (d, 2H, J = 8.4 Hz, Ar), 5.6 (s, 2H, CH2OAr)5.17 (s, 2H, ArCH2N), 4.83 (d, 2H, J = 6.0 Hz, HNCH2Ar); 13C NMR (DMSO-d6, 100 MHz): δ 177.3 (C=S), 159.4, 139.6, 136.0, 128.9, 128.8, 128.2, 128.1, 128.0, 127.2, 127.1, 126.7, 114.9 (Ar), 142.8 (N-C=CHN), 142.1 ArCH = N), 124.8 (N-C = CHN), 61.2 (ArCH2N), 52.9 (CH2OAr)46.5 (HNCH2Ar); Anal. Calcd. for C25H24N6OS: C, 65.77; H, 5.30; N, 18.40; S, 7.02. Found: C, 65.83; H, 5.82; N, 18.38; S, 6.74 %.
(Z)-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy]benzylidene}-N-phenethylhydrazinecarbothioamide (1h): 74 % yield; white solid; mp 165-167 °C; IR (neat, cm-1): 3298, 3149,1602,1298,1223,1049 and 825; 1H NMR (CDCk, 400 MHz): δ 9.69 (bs, 1H, NH), 7.74 (s, 1H, ArCH=N), 7.52 (s, 1H, H-triazole), 7.42 (d, 2H, J = 8.4 Hz, Ar),7.34-7.24(m,10H,Ar),6.94(d,2H, J = 8.4 Hz, Ar), 5.52(s,2H, CH2OAr), 5.20 (s, 2H, ArCH2N), 4.02-3.84 (m, 2H, HNCH2CH2Ar), 2.96 (t, 2H, J = 6.8 Hz, HNCH2CH2Ar); 13C NMR(CDCl3, 100 MHz): δ 177.0 (C=S), 160.0, 138.6, 134.3, 129.1, 128.9, 128.8, 128.7, 128.1, 126.6, 126.5, 115.1 (Ar), 144.0 (N-C=CHN), 142.0 (ArCH=N), 122.7 (N-C=CHN), 62.1 (ArCHN), 54.3 (CH2OAr), 45.3 (HNCH2CH2Ar), 35.3 (HNCH2CH2Ar); Anal. Calcd. for C26H26N6OS: C, 66.36; H, 5.57; N, 17.86; S, 6.81 %. Found: C, 65.72; H, 5.61; N, 17.73; S, 7.22 %.
(Z)-N-anilinyl-2-{4-[(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy] benzylidene}-N-phenethylhydrazinecarbothioamide (Li) 64 % yield; grey solid; mp 167-169 °C; IR (neat, cm-1): 3299,3235,3135,3048, 1603, 1289, 1225, 1054, 822; 1H NMR: (DMSO-d6, 400 MHz): δ 10.13 (bs, 1H, NH), 8.26 (s, 1H, H-triazole), 7.80 (s, 1H, ArCH=N), 7.56 (d, 2H, J = 8.4 Hz, Ar), 7.43-6.61 (m, 12H, Ar), 5.59 (s, 2H, CH2OAr), 5.14 (s, 2H, ArCH2N); 13C NMR: (DMSO-d6, 100 MHz): δ 158.2, 136.6, 136.1, 129.3, 129.0, 128.9, 128.4, 128.1, 127.2, 118.6, 115.2, 112.0 (Ar), 145.7 (N-C = CHN), 143.1 (ArCH=N), 124.9 (N-C=CHN), 61.2 (ArCH2N), 53.0 (CH2OAr); Anal. Calcd. for C24H23N7OS : C, 63.00; H, 5.07; N, 21.43; S, 7.01 %. Found: C, 62.88; H, 5.17; N, 21.17; S, 6.96 %.
Antimalarial Assay
The in vitro antimalarial activity of the hybrids 1a-i against the 3D7 strain of the malaria parasite, Plasmodium falciparum,is measured by assessing parasite survival after drug exposure using a parasite lactate dehydrogenase (pLDH) colorimetric enzyme assay. Lactate dehydrogenase is an enzyme found in all cells and catalyses the formation of pyruvate from lactate by reducing the co-enzyme NAD + (nicotinamide adenine dinucleotide) to NADH. In the pLDH assay, the NAD analogue APAD (3-acetylpyridine adenine nucleotide) is reduced to APADH and in turn a yellow NBT/PES (nitro blue tetra-zolium + phenazine ethosulphate) reagent is converted to purple formazan crystals. The absorbance is read at 620 nm using a multi-well spectrophotometer (Tecan Infinite F500). Formazan formation is directly proportional to pLDH activity which in turn is indicative of the number of parasites in the cultures following drug exposure. Assay specificity is ensured by the inability of human LDH, found in the host red blood cells, to use APAD as a co-factor.
Compound inhibitory activity is determined by preparing test samples in parasite culture medium in transparent 96-well flat bottom plates (Greiner Bio-one) - 10 concentrations (n = 4 for each data point). Parasitized red blood cells are added to a final concentration of 1 % haematocrit, 2 % parasitaemia and the plates incubated for 48 hours before proceeding with the pLDH assay. Percentage parasite survival in each well is calculated relative to control wells that receive no drug.
Crystal Structure Report
A colourless, plate-like specimen of C17H17N3O2, approximate dimensions 0.05 mm x 0.20 mm x 0.25 mm, was used for the X-ray crystallographic analysis. The X-ray intensity data were measured as follows. A total of 2980 frames were collected. The total exposure time was 8.28 hours. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The integration of the data using an orthorhombic unit cell yielded a total of 26836 reflections to a maximum è angle of 65.49° (0.85 A resolution), of which 2479 were independent (average redundancy 10.825, completeness = 99.2 %, Rint = 3.52%,Rsig = 1.77%)and2333 (94.11 %) weregreaterthan2σ(F2). The final cell constants of a = 11.2999(4) Å, b = 8.3445(3) Å, c = 30.7514(10) Å, volume = 2899.61(17) Å3, are based upon the refinement of the XYZ-centroids of 9837 reflections above 20 σ(Γ) with 5.748 ° < 2θ < 130.1 °. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.827. The calculated minimum and maximum transmission coefficients (based on crystal size) are 0.8397 and 0.9655. The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group Pbca,with Z = 8 for the formula unit, C17H17N3O2. The final anisotropic full-matrix least-squares refinement on F2 with 199 variables converged at R1 = 4.08 %, for the observed data and wR2 = 10.68 % for all data. The good-ness-of-fit was 1.222. The largest peak in the final difference electron density synthesis was 0.139 e-/Å3 and the largest hole was -0.675 e-/Å3 with an RMS deviation of 0.047 e-/Å3. On the basis of the final model, the calculated density was 1.353 g/cm3 and F(000), 1248 e-. Crystallographic data for the structure have been deposited with the Cambridge Crystallographic Data Centre as deposition No. CCDC-934893.
Acknowledgements
We would like to acknowledge the University of Johannesburg for financial support and the XRD unit in the department of Chemistry, especially Mrs Z. Phasha, for solving the crystal structure of triazole 4. The Biosciences and Biomedical Technologies research group of the CSIR are also acknowledged for testing our compounds for their antimalarial activity.
References
1 World Health Organization, 2000, WHO Expert Committee on Malaria: Twentieth Report, Technical Report 892, Geneva. [ Links ]
2 S. Vangapandu, S. Sachdeva, M. Jain, S. Singh, P.P. Singh, C.L. Kaul and R. Jain, Bioorg. Med. Chem. 2003, 11, 4557-4568. [ Links ]
3 V.V Kouznetsov and A. Gómez-Barrio, Eur. J. Med. Chem., 2009, 44, 3091-3113. [ Links ]
4 R. Capela, R. Oliveira, L.M. Goncalves, A. Domingos, J. Gut, P.J. Rosenthal, F. Lopes and R. Moreira, Bioorg. Med. Chem. Lett., 2009, 19, 3229-3232. [ Links ]
5 N.C. Araújo, V. Barton, M. Jones, P.A. Stocks, S.A. Ward, J. Davies, P.G. Bray, A.E. Shone, M.L. Cristiano and P.M. O'Neill, Bioorg. Med. Chem. Lett., 2009, 19, 2038-2043. [ Links ]
6 R.J. Maude, C.J. Woodrow and L.J. White, Drug Dev. Res., 2010, 71, 12-19. [ Links ]
7 H. Noedl, Y. Se, K. Schaecher, B.L. Smith, D. Socheat and M.M. Fukuda, N. Engl. J. Med., 2008, 359, 2619-2620. [ Links ]
8 L.K. Basco, O. Dechy-Cabaret, M. Ndounga, F.S. Meche, A. Robert and B. Meunier, Antimicrob. Agents CH., 2001, 45, 1886-1888. [ Links ]
9 R. Morphy, C. Kay and Z. Rankovic, Drug Discovery Today, 2004, 9, 641-651. [ Links ]
10 C. Loup, J. Lelievre, F. Benoit-Vical and B. Meunier, Antimicrob. Agents CH., 2007, 51, 3768-3770. [ Links ]
11 F. Coslédan, L. Fraisse, A. Pellet, F. Guillou, B. Mordmuller, P.G. Kremsner, A. Moreno, D. Mazier, J.P. Maffrand and B. Meunier, Proc. Natl. Acad. Sci. USA., 2008, 105, 17579-17584. [ Links ]
12 C. Shipman Jr, H. Smith, J.C. Drach and D.L. Klayman, Antiviral Res., 1986, 6,197-222. [ Links ]
13 V. Philip,V Suni, M.R.P. Kurup and M. Nethaji, Polyhedron, 2006, 25, 1931-1938. [ Links ]
14 A. Usman, L.A. Razak, S. Chantrapromma, H.K. Fun, A. Sreekanth, S. Sivakumar and M.R.P. Kurup, Acta Cryst. C, 2002,461. [ Links ]
15 H.C. KolbandK.B. Sharpless, DrugDiscoveryToday,2003, 8, 1128-1137. [ Links ]
16 Y. Zou, Q. Zhao, J. Liao, H. Hua, S. Yu, X. Chai, M. Xu and Q. Wua, Bioorg. Med. Chem. Lett., 2012, 22, 2959-2962. [ Links ]
17 S.E. Livingstone, Quart. Rev. Chem. Soc., 1965, 19, 386-425. [ Links ]
18 B.B. Modzelewska and W.E. Jagiello, Acta Pol. Pharm., 2000, 57, 199-204. [ Links ]
19 J.Y. Jin, L.X. Zhang, X.X. Chen, A.J. Zhang and H.L. Zhang, Molecules, 2007, 12, 297-303. [ Links ]
20 S. Hoogendoorn, A.E.M. Blom, L.I. Willems, G.A. van der Marel and H.S. Overkleeft, Org. Lett., 2011, 13, 5656-5659. [ Links ]
21 W.L. Jae, K.K. Byung, H.K. Jung, S.S. Won and H.J. Sung, Bull. Korean. Chem. Soc., 2005, 26, 1790-1794. [ Links ]
22 C. Bhadaliya, B. Mahesh, J. Monika, N.S. Barij, S. Philip, G. Geraldine, S. Thomas, R. Ganapathy, R. Praveen, M. Dhatchana, B. Arijit and J. Venkatesan, Bioorg. Med. Chem. Lett., 2010, 20, 3906-3910. [ Links ]
23 D.L. Klayman, J.F. Bartosevich, T.S. Griffin, C.J. Mason and J.P. Scovill, J. Med. Chem., 1979, 22, 855-862. [ Links ]
24 R.H. Hans, E.M. Guantai, C. Lategan, P.J. Smith, B. Wanc, S.G. Franzblau, J. Gut, P.J. Rosenthal and K. Chibale, Bioorg. Med. Chem. Lett., 2010, 20, 942-944. [ Links ]
25 M.A. Garcia, Z.G. Rios, J. Gonzalez, V.M. Perez, N. Lara, A. Fuentes, C. Gonzalez, D. Corona and E. Cuevas-Yanez, Lett. Org. Chem., 2011, 8, 701-706. [ Links ]
Received 14 March 2013
Revised 10 April 2013
Accepted 23 April 2013
* To whom correspondence should be addressed. E-mail: hhkinfe@uj.ac.za

