










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
RESEARCH ARTICLE
Synthesis of Alkyne and Alkene Ketal Derivatives of Pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8-11-dione and 1-Phenyl-pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8-11-dione
Frans T.I. Marx; Johan H.L. Jordaan; Mariké Nel; Hermanus C.M. Vosloo
Research Focus Area for Chemical Resource Beneficiation: Catalysis and Synthesis Research Group, North-West University, Potchefstroom, 2520, South Africa
ABSTRACT
Functionalizingpentacyclo[5.4.0.02,6.03,10.05,9]undecane-8-11-oneand 1-phenyl-pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8-11-one can be easily accomplished by using the alcohols of various alkynes and alkenes. Generally the synthesis of terminal alkyne and cyclic alkene ketal derivatives were performed fairly easily. Synthesis of the terminal alkenes provided some difficulties. Reduction of the alkyne ketal derivatives using Pd/CaCO3 has been demonstrated to form a mixture containing the desired alkene ketal derivatives.
Keywords: Cage compounds, diketone, ketal formation, reduction.
1. Introduction
The synthesis and chemistry of alicyclic compounds with cage structures has been the subject of research by many groups over the past 50 years.1 These cage structures have found applications in medicine where they, for instance enhance transport over mem-branes.2 A key step in the synthesis of such compounds are the photochemical cyclisation of a Diels-Alder adduct. The adduct of p-benzoquinone and cyclopentadiene has a structure with two double bonds within reach of each other in the same molecule, which undergoes photochemical addition to yield the highly strained dione 1.3 In a similar manner the dione 2 can be synthe-sized by starting with 2-phenyl-1,4-benzoquinone.4
In literature only a few examples exist of the synthesis of alkyne and alkene derivatives of 1.5-8 Functionalizing these diones with alkynes and alkenes provides many new possible substrates for inter alia alkyne and alkene metathesis reactions as well as monomers for polymerisation reactions. In this paper we report the synthesis of alkyne ketal derivatives of different chain lengths as well as linear alkene and cyclic alkene ketal deriva-tives. Reduction of the alkyne ketal derivatives with Pd/CaCO3 has been done to form a mixture containing the desired alkene ketal derivatives.
2. Results and Discussion
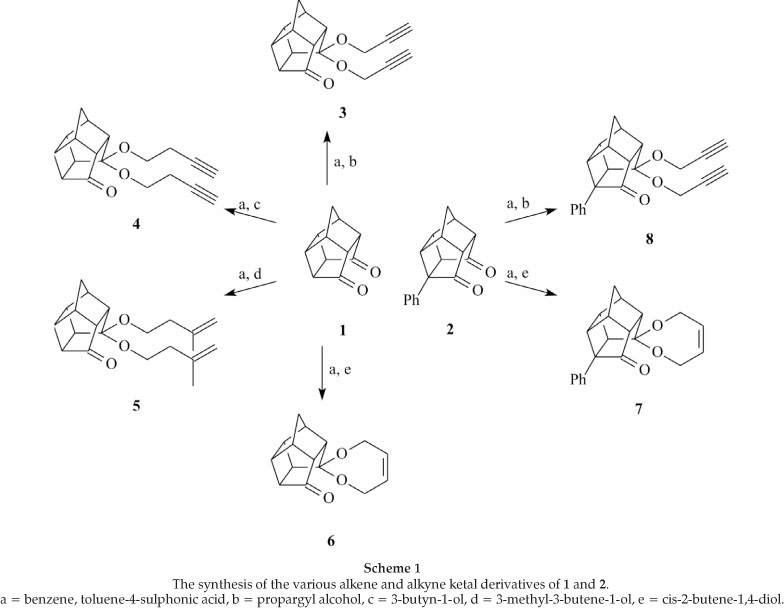
We found that the dione 1 gives various alkynes (3 and 4) and alkene 5 as well as the cyclic alkene 6 when reacted with various alkyne and alkene alcohols respectively in the presence of a catalytic amount of acid. When the dione 2 was used as the starting material instead of 1 similar results could be achieved (7 and 8). The yields for all the reactions are listed in Table 1. During our investigation we found that the terminal alkyne and cyclic alkene syntheses produced the easily isolatable crystalline prod-ucts 3, 4, 6, 7 and 8. The alkene 5, which contains terminal alkenes, is obtained as light yellow oil. During the synthesis of 5 we found that a high yield could be achieved in one hour, but unwanted by-products formed in low yields, which could only be separated with tedious column chromatography separations. If the reaction was refluxed overnight no 5 could be isolated. We were unable to separate the by-products sufficiently for NMR analyses. Analysis by GC/MS indicated three major by-products that could not be identified unambiguously. In none of the other syntheses these by-products were observed. After several months the isolated compound 5 remained stable; no decomposition products were observed. This indicates that the by-product formation is dependent on the reaction conditions used during synthesis.

Although it is not indicated in the various schemes in this paper, all the products were racemic mixtures of equal distribution. The only exceptions were 7 and 8 where the main products were those indicated in the schemes. The ketal groups and the phenyl group are on opposite sides of the cage compound. The ratios of the racemic mixtures determined via GC-MS analysis were 9:1 for 7 and for 8 it was 30:1.

Recently Kotha et al.9,10 reported the ring-closing metathesis (RCM) of 9 with various Grubbs-type precatalysts in toluene to yield 10 in a high yield of 87 %.
In order to synthesize reagents for similar alkene metathesis reactions we performed alkyne to alkene reductions with Lindlar's11 catalyst, Pd/CaCO3, in a hydrogen atmosphere on 3 and 4. In both cases a mixture of products were isolated in moderate yields as light yellow oils. GC-MS analyses of the oils showed a single broad peak for compounds 11 and 12, the ratio for this mixture to 13 were 76:23 and the ratio for 14,15 and 16 were 35:35:30.
Complete conversion of the alkyne derivatives 3 and 4 were obtained as no alkyne CH- or C≡C-stretch absorption bands were observed in the IR-spectra of the resulting mixtures. Furthermore no molecular masses for 3 and 4, or the mono-alkyne derivatives 17 and 18 were observed using GC-MS after isolation. We were not able to separate compounds 11 to 16 from their respective mixtures. The complete reduction of 3 to 13 with 10 % Pd/C in a hydrogen atmosphere was performed to give an isolated yield of 93 %. Using the NMR data from 13 we were able to assign the NMR spectra of the mixtures of 11, 12, and 13, and 14, 15 and 16' respectively.
During the reduction of 3 with Lindlar's catalyst the product formation was as follows: 3 → 19 → 11 → 12 → 13. The reduction of 4 followed the route: 4 → 20 → 14 → 15 → 16. If the formation of 17 or 18 takes place during the reduction, it is in such a small quantity that we were unable to isolate it. Although we chose a side at random in the schemes to illustrate the reduction process, we were not able to determine if reduction will take place first on the endo or on the exo branch of the ketal substituents.
3. Conclusions
The various synthesis methods discussed provide an easy and efficient way to synthesize various alkyne, alkene and cyclic alkene moieties of the diones 1 and 2 that can potentially be used as monomers for polymerisation and metathesis reactions.
4. Experimental
General methods
All reactants and solvents were used as received from the sup-pliers (Fluka, Adrich and Merck). All the diene- and diyne alco-hols were distilled before being used. Infrared spectra (KBr-disk) were recorded on a Nicolet 550 Magna IR spectrometer and on a Bruker Alpha P ATR. EI spectra were obtained at 70 eV on a Micromass Autospec-Tof- and a Thermo DFS magnetic sector mass spectrometer (HRMS-EI). Reactions were monitored with an Agilent 6890 gas chromatograph equipped with an Agilent 7683 autosampler, HP-5 capillary column and an Agilent 5973 mass selective detector (MSD). 300 MHz 1H NMR and 75 MHz 13C NMR spectra were recorded on a Varian Gemini-300 spectrometer and 600 MHz 1H NMR and 150 MHz 13C NMR spectra were recorded on a Bruker Avance III UltraShield Plus 600 MHz spectrometer. All melting points were determined with a Buchi B-540 apparatus.
General procedure for the synthesis of alkene and alkyne ketals
In a Dean-Stark apparatus the dione, the alcohol and a cata-lytic amount of toluene-4-sulphonic acid was dissolved in ben-zene. The reaction mixture was refluxed for 1 to 24 hours. In some cases more alcohol was added to ensure the complete conversion of the substrate to product. The water that distilled off, very fast initially, was removed to help ensure that the product formation was maximized. The reaction was quenched by adding a 10 % sodium carbonate solution to neutralize the acid. Extraction was done with3x10mL dichloromethane. The combined organic layers were washed once with water, dried over magnesium sulfate and filtered.
Pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,8-diprop-2-ynyloxy-11-one (3): 1 (10.03 g, 57.6 mmol), propargyl alcohol (6.74 g, 120.2 mmol) and toluene-4-sulphonic acid (0.41 g, 2.4 mmol) were dissolved in 100 mL benzene and refluxed for 3 hours where after propargyl alcohol was added (5 mL, 85.9 mmol). After another 3 hours another portion propargyl alcohol was added (15 mL, 257.7 mmol) and the reaction was refluxed for 16 hours where af-ter the reaction was stopped and isolated as described above. A pure product of 3 was obtained by recrystallization from abso-lute ethanol (12.84 g, 83 %, mp 122 °C) as an off-white powder. IR (KBr-disk): vmax 3281, 3250, 2996, 2929, 2914, 2859, 2140,1734, 1460, 1390, 1359, 1109, 1085, 1062, 1046, 687, 640 cm-1. HRMS-EI, m/z calc: 268.1099, found: 268.1096 [M+]. 13C [(CD3)2SO], δC 76.52 (D), 76.34 (D), 51.54 (T), 51.21 (D), 49.55 (T), 48,97 (D), 45,29 (D), 42.05 (D), 41.23 (D), 41.14 (D), 40.00 (D), 37.57 (T), 35.43 (D). 1H [(CD3)2SO], δH 4.16-4.09 (dd, 2xH), 4.04-3.96 (dd, 2xH), 3.48-3.36 (t, 1xH), 3.36-3.32 (t, 1xH), 3.06-2.79 (m, 3xH), 2.79-2.66 (m, 2xH), 2.57-2.46 (m, 1xH), 2.44-2.33 (m, 1xH), 3.32-2.16 (dp, 1xH), 1.89-1.68 (d, JAB = 10.71 Hz ½ x CH2) and 1.55-1.36 (d, JAB = 10.79 Hz ½ x CH2).
Pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,8-dibut-3-ynyloxy-11-one (4): 1 (4.01 g, 23.0 mmol), 3-butyn-1-ol (3.90 g, 55.6 mmol) and toluene-4-sulphonic acid (0.20 g, 1.2 mmol) were dissolved in 100 mL benzene and refluxed for 1 hour where after 3-butyn-1-ol was added (1 mL, 13.2 mmol). After 3 hours the reaction was stopped and isolated as described above. A pure product of 4 was obtained by recrystallization from absolute ethanol (6.56 g, 96 %,mp 103 °C) as an off-white powder. IR (KBr-disk): vmax 3259, 2984, 2960, 2934, 2887, 2868, 2116, 1730, 1452, 1392, 1327, 1173, 1165,1109,1096,1033,749,680 cm-1. HRMS-EI, m/z calc: 296.1099, found: 296.1096 [M+]. 13C [(CD3)2SO], δC 71.86 (D), 71.59 (D), 61.83 (T), 59.42 (T), 51.27 (D), 49.03 (D), 45.13 (D), 42.10 (D), 41.31 (D), 41.26 (D), 40.02 (D), 37.69 (T), 35.49 (D), 19.47 (T), 18.60 (T). 1H [(CD3)2SO], δH 3.62-3.43 (m, 2xH), 3.38-3.25 (t, 2xH), 2.96-2.76 (m, 3xH), 2.76-2.73 (m, 3xH), 2.72-2.64 (t, 1xH), 2.55-2.47 (t, 1xH), 2.45-2.32 (m, 3xH), 2.32-2.14 (m, 3xH), 1.85-1.68 (d, JAB = 10.85 Hz½xCH2) and 1.58-1.37 (d, JAB = 10.71 Hz ½ x CH2).
Pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,8-di(3-methyl-but-3-enyloxy)-11-one (5): 1 (5.05 g, 29.0 mmol), 3-methyl-3-butene-1-ol (15.82 g, 183.7 mmol) and toluene-4-sulphonic acid (0.21 g, 1.2 mmol) were dissolved in 100 mL benzene and refluxed for 1 hour where after the reaction was stopped and isolated as described above. A pure product of 5 was obtained by perform-ing flash chromatography (2:1 petroleum ether (60-80 °C):ethyl acetate) (7.05 g, 74 %)asa light yellow oil. IR (KBr-disk): vmax 3075, 2971,2935,2870,1745,1649,1444,1327,1172,1108,1050,987,941, 748 cm-1.EIMS, m/z calc: 328.2038, found: 328.2049 [M+]. 13C [(CD3)2SO], δC 111.39 (T), 111.36 (T), 61.81 (T), 59.24 (T), 51.34 (D), 49.02 (D), 45.22 (D), 42.04 (D), 41.23 (D), 40.04 (D), 37.66 (T), 37.61 (T), 36.79 (T), 35.46 (D), 22.37 (Q). 1H [(CD3)2SO], δH 4.86-4.52 (m, 4xH), 3.53-3.39 (m, 2xH), 3.32-3.25 (m, 2xH), 3.01-2.66 (m, 4xH), 2.66-2.58 (t, 1xH), 2.55-2.44 (t, 1xH), 2.44-2.31 (m, 1xH), 2.31-2.02 (m, 5xH), 1.73-1.639 (d, 6xH), 1.82-1.73 (d, JAB = 10.72Hz½xCH2) and1.54-1.40 (d,JAB = 10.71 Hz½xCH2). Pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,8-but-2-ene-1,4-dioxy-11-one (6): 1 (0.18 g, 1.0 mmol), cis-2-butene-1,4-diol (0.11 g, 1.2 mmol) and toluene-4-sulphonic acid (0.03 g, 0.2 mmol) were dissolved in 40 mL benzene and refluxed for 3.5 hours where after the reaction was stopped and 6 was isolated as described above. A pure product of 6 was obtained by washing the product through silica gel (1:1 petroleum ether (60-80 °C):ethyl acetate) (0.23 g, 91 %, mp 117 °C) as an off-white powder. IR (KBr-disk): vmax 3032,2972,2951,2935,2864,1738,1451,1384,1334,1197,1161, 1110, 1085, 767 cm-1. HRMS-EI, m/z calc: 244.1094, found: 244.1091 [M+]. 13C [(CD3)2SO],δC 129.36 (D), 128.92 (D), 63.87(T), 61.16 (T), 50.56 (D), 49.34 (D), 45.50 (D), 42.03 (D), 41.20 (D), 40.39 (D), 40.21 (D), 37.88 (T), 35.43 (D). 1H [(CD3)2SO], dH 5.80-5.45 (q, 2xH), 4.29-3.90 (dd, 4xH), 3.05-2.69 (m, 4xH), 2.69-2.60 (dd, 1xH), 2.55-2.45(dd, 1xH) 2.44-2.33 (dt, 1xH), 2.32-2.16 (dt, 1xH), 1.87-1.68 (d, JAB = 10.74 Hz½xCH2) and 1.59-1.36 (d, JAB = 10.71 Hz ½ x CH2).
1-Phenyl-pentacyclo[5A.0.02",6.03,10.05,9]-undecane-8,8-but-2-ene-dioxy-11-one (7): 2 (0.25 g, 1.4 mmol), cis-2-butene-1,4-diol (0.15 g, 1.7 mmol) and toluene-4-sulphonic acid (0.03 g, 0.2 mmol) were dissolved in 40 mL benzene and refluxed for 3 hours where after the reaction was stopped and isolated as described above. A pure product of the two possible isomers of 7 was obtained by washing the product through silica gel (1:1 petroleum ether (60-80 °C):ethyl acetate) (0.21 g, 66 %, mp 121 °C) as an off-white powder. IR (KBr-disk): νmax 3091, 3060, 3048, 3031, 3001, 2981, 2967, 2946, 2924, 2866, 1737, 1606, 1499, 1446, 1390, 1172, 1116, 1091, 1082, 1024, 1006, 937 744, 696 cm-1. HRMS-EI, m/z calc: 320.1407, found: 320.1396 [M+]. 13C [(CD3)2SO], δC 129.45 (D), 128.88 (D), 127.83 (D), 127.22 (D), 126.05 (D), 63.98 (T), 61.38 (T), 50.48 (D), 50.42 (D), 47.98 (D), 45.34 (D), 42.45 (D), 41.43 (D), 39.28 (D), 38.24 (T). 1H [(CD3)2SO], dH 7.44-7.28 (m, 2xH), 7.27-7.21 (dd, 1xH), 7.20-7.01 (m, 2xH), 5.81-5.54 (t, 2xH), 4.40-4.05 (t, 4xH), 3.10-3.00 (m, 1xH), 3.00-2.84 (m, 3xH), 2.79-2.62 (dt, 2xH), 2.54-2.40 (m, 1xH), 1.96-1.76 (d, JAB = 10.74 Hz ½ x CH2) and 1.70-1.47 (d, JAB = 10.74 Hz ½ x CH2).
1-Phenyl-pentacyclo[5.4.0.02,6.03,10.05,9]-undecane-8,8-diprop-2-ynylo xy-11-one (8): 2 (0.25 g, 1.4 mmol), propargyl alcohol (0.15 g, 2.7 mmol) and toluene-4-sulphonic acid (0.01 g, 0.1 mmol) were dissolved in 30 mL benzene and refluxed for 20 hours where after the reaction was stopped and isolated as described above. A pure product of the two possible isomers of 8 was obtained by performing flash chromatography (1:1 petroleum ether (60-80 °C):ethyl acetate) (0.12 g, 35 %, mp 102 °C) as an off-white powder. IR (KBr-disk): νmax 3265, 3050, 3020, 2970, 2920, 2860, 2120,1740,1610,1450,1400,1120,1090,1080,1050,1015,780,700, 620 cm-1. HRMS-EI, m/z calc: 344.1407, found: 344.1399 [M+]. 13C [(CD3)2SO], δC 127.87 (D), 127.26 (D), 126.16 (D), 76.70 (D), 76.48 (D), 52.00 (T), 51.82 (D), 50.25 (D), 49.88 (T), 47.58 (D), 45.56 (D), 43.12 (D), 41.43 (D), 39.17 (D), 38.00 (T). 1H [(CD3)2SO], δH 7.43-7.28 (t, 2xH), 7.28-7.10 (m, 3xH), 4.24-4.19 (d, 1xH), 4.19-4.04 (dd, 1xH) 3.41-3.35 (m, 2xH), 3.23-3.13 (m, 1xH), 3.05-2.87 (m, 3xH), 2.84-2.75 (t, 1xH), 2.75-2.63 (t, 1xH), 2.55-2.35 (m, 1xH), 1.90-1.81 (d, JAB = 10.79 Hz½xCH2) and 1.68-1.49 (d, JAB = 10.82 Hz ½ x CH2).
General procedure for the Lindlar reduction of the alkyne ketals11
In a round-bottom flask maintained under a hydrogen atmo-sphere Lindlar's catalyst (10 % Pd/CaCO3), quinoline and the alkyne ketal were dissolved in acetone. The reaction mixture was kept under hydrogen until a satisfactory conversion to the alkene-ketal was achieved. The product was purified by performing flash chromatography (1:1 petroleum ether (60-80 °C):ethyl acetate).
Pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,8-diprop-2-enyloxy-11-one (11): 3 (0.54 g, 2.0 mmol), Lindlar's catalyst (0.045 g, 0.04 mmol Pd, 0.41 mmol CaCO3) and quinoline (0.1 mL, 0.85 mmol) were dissolved in 70 mL acetone and stirred under hydrogen for 18 h after which the reaction was stopped and a mixture of 11,12 and 13 (0.27 g, 50 %) was obtained by isolation as described above as a yellow oil. IR (KBr-disk): νmax 3078, 2968, 2929, 2851, 1746, 1615, 1460, 1429, 1331, 1178, 1115, 1070, 992, 937 cm-1. GC-MS, 11 m/z 272 [M+], 12 m/z274 [M+] and 13 m/z276 [M+]. 13C [(CD3)2SO], δC 135.27 (D), 134.25 (D), 116.25 (D), 116.07 (D), 115.56 (d), 64.5 (T), 64.27 (T), 62.14 (T), 61.82 (T), 51.37 (D), 51.28 (D), 49.01 (D), 48.98 (D), 45.25 (D), 45.18 (D), 42.02 (D), 41.25 (D), 40.00 (D), 37.63 (T), 35.45 (D), 22.61 (T), 10.58 (Q), 10.53 (Q). 1H [(CD3)2SO], δH 5.98-5.65 (m, 2xH), 5.30-5.12 (m, 2xH), 5.12-4.99 (m, 2xH), 3.97-3.83 (m, 3xH), 3.83-3.68 (m, 1xH), 3.39-3.22 (m, 2xH), 3.02-2.77 (m, 5xH), 2.77-2.69 (m, 2xH), 2.68-2.56 (m, 2xH), 2.56-2.43 (t, 2xH), 2.43-2.31 (m, 2xH), 2.31-2.17 (m, 2xH), 1.55-1.30 (m, 2xH), 0.92-0.71 (p, 3xH), 1.86-1.69 (d, JAB = 10.71 Hz ½ x CH2) and 1.55-1.30 (d, JAB = 10.65 Hz ½ x CH2).
Pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,8-di(3-methyl-but-3-enyloxy)-11-one (14): 4 (1.10 g, 3.7 mmol), Lindlar's catalyst (98.8 mg, 0.09 mmol Pd, 0.89 mmol CaCO3) and quinoline (0.2 mL, 1.69 mmol) were dissolved in 70 mL acetone and stirred under hydrogen for 40 h whereafter the reaction was stopped and a mixture of 14,15 and 16 (928 mg, 83 %) was obtained by isolation as described above as an yellow oil. IR (KBr-disk): νmax 2959,2933, 2869, 1736, 1641, 1328, 1106, 1067, 1050, 942, 911, 826 cm-1. HRMS-EI, 14 m/z calc: 300.1719, found: 300.1714 [M+], 15 m/z calc: 302.1876, found: 302.1877 [M+] and 16 m/z calc: 304.2038, found: 304.2033 [M+]. 13C [CDCl3], δC 135.27 (D), 135.23 (D), 135.10 (D), 135.06 (D), 116.36 (D), 116.31 (D), 116.26 (D), 63.66 (t), 63.57 (T), 63.30 (T), 63.22 (T), 60.89 (T), 60.81 (T), 60.67 (T), 60.60 (T), 52.16 (D), 52.12 (D), 52.11 (D), 49.69 (D), 45.70 (D), 42.74 (D), 42.03 (D), 41.99 (D), 41.78 (D), 40.77 (D), 38.67 (T), 38.20 (T), 36.16 (D), 34.40 (T), 34.35 (T), 33.61 (T), 33.58 (T), 32.07 (T), 32.03 (T), 31.39 (T), 31.36 (T), 19.45 (T), 19.43 (T), 19.35 (T), 13.86 (Q). 1H [CDCl3], δH 6.44-5.51 (m, 2xH), 5.17-4.55 (m, 2xH), 4.55-3.08 (m, 2xH), 3.08-2.81 (m, 2xH), 2.81-2.62 (m, 2xH), 2.62-2.50 (m, 1xH), 2.50-2.39 (m, 1xH), 2.39-2.26 (m, 1xH), 2.26-1.97 (m, 1xH), 1.48-1.04 (m, 2xH), 1.04-1.09 (dt, 3xH), 1.97-1.65 (d, JAB = 10.56 Hz½xCH2) and 1.65-1.48 (d, JAB = 10.92 Hz ½ x CH2).
Pentacyclo[5.4.0.02,6.0s,10.05,9]undecane-8,8-dipropyloxy-11-one (13): 3 (270 mg, 1.0 mmol) and 10 % Pd/C (2.0 mg, 1.88 jumol Pd, 0.15 mmol C) were dissolved in 70 mL acetone and stirred under hydrogen for 72 h where after the reaction was stopped and 13 (258 mg, 93 %) was obtained as an colourless oil. IR (KBr-disk): νmax 2968, 2929, 2876, 1746, 1484, 1392, 1337, 1160, 1123, 1076, 1060 cm-1. HRMS-EI, m/z calc: 276.1715, found: 276.1719 [M+]. 13C [(CD3)2SO], δC 64.64 (T), 61.99 (T), 51.34 (D), 48.97 (D), 45.19 (D), 42.02 (D), 41.32 (D), 41.22 (D), 40.02 (D), 37.65 (T), 35.45 (D), 22.69 (T), 22.00 (T), 10.64 (Q), 10.58 (Q). 1H[(CD3)2SO], δH 3.42-3.22 (m, 4xH), 3.03-2.81 (m, 2xH), 2.81-2.67 (m, 2xH), 2.67-2.56 (m, 1xH), 2.56-2.44 (m, 1xH), 2.44-2.31 (m, 1xH), 2.31-2.16 (dp, 1xH), 1.58-1.31 (m, 4xH), 0.95-0.72 (dd, 6xH) 1.86-1.70 (d, JAB = 10.72 Hz/xCH2) and 1.49-1.42 (d, JAB = 10.82 Hz ½ x CH2).
Acknowledgements
We would like to thank the Research Focus Area for Chemical Resource Beneficiation at the North-West University for providing the necessary funding and laboratory facilities to complete this work. We would also like to thank Mr. André Joubert for his valuable help in recording the NMR spectra.
References
1 G.W. Griffin & A.P. Marchand, Chem. Rev., 1989, 89, 997-1010. [ Links ]
2 J.L. Neumeyer & R.G. Booth, Neuroleptics and anxiolitic agents, in Principles of Medicinal Chemistry, (W.O. Foye, T.L. Lemke & D.A. Williams, eds.), 4th edn., Lea & Febiger, London, 1989, 199-231. [ Links ]
3 R.C. Cookson, E. Crundwell, R.R. Hill, J.J. Hudec, Chem. Soc., 1964, 3062-3075. [ Links ]
4 A.P. Marchand, P. Annapurna, S. Pulla Reddy, W.H Watson & A.J. Nagl, Org. Chem., 1989, 54, 187-193. [ Links ]
5 A.P. Marchand, K.A. Kumar & A.S. Mckim, Tetrahedron, 1997, 53, 3467-3474. [ Links ]
6 S.G. Bott, A.P. Marchand & K.A. Kumar, J. Chem. Crystallogr, 1999,29, 347-350. [ Links ]
7 A.A. Pinkerton, M.J. Hardie, A.P. Marchand & K.A. Kumar, J. Chem. Crystallogr., 2001, 135-142. [ Links ]
8 S. Kotha, E. Manivannan & N. Sreenivasachary, J. Chem. Soc., Perkin Trans. 1, 1999, 2845-2848. [ Links ]
9 S. Kotha & M.K.Dipak, Chem. Eur. J., 2006,12, 4446-4450. [ Links ]
10 S. Kotha, V Seema, K. Singh, K.D. Deodhar, Tetrahedron Lett, 2010,51, 2301-2304. [ Links ]
11 H. Lindlar & R. Dubuisin, Paladium catalyst for partial reductionof acetylenes, in Organic Synthesis, (H.E. Baumgarten, ed.), vol. V, John Wiley and Sons, New York, 1973, pp. 880-883. [ Links ]
 Correspondence:
Correspondence:
E-mail: johan.jordaan@nwu.ac.za
Received 26 August 2013
Revised 25 September 2013
Accepted 4 November 2013


{kind=link}
{kind=link}