










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
RESEARCH ARTICLE
Conformational preferences of Amphibian peptides Brevinin-Ya and Brevinin-Yb explored using molecular dynamics
Parvesh Singh*
Department of Chemistry. Durban University of Technology, Durban 4000, South Africa
ABSTRACT
The brevinin-1 family peptides obtained from different frog skins have great potency against bacterial and fungal infections. Their biological activities are significantly affected by mutations especially at positions 11 and 14. However, despite having great medicinal potential, the detailed information regarding their three-dimensional structures is still not fully known. In the present study, the conformational profiles of two brevinin peptides (Brevinin 1-Ya and Brevinin 1-Yb) were explored at molecular mechanics level using molecular dynamics (MD) method. Specifically, four MD simulations (Brev1_Ya(E), Brev1_Yb(E), Brev1_Ya(H), Brev1_Yb(H)) were performed starting from extended as well as helical conformations of both peptides under implicit solvent conditions. The analysis of the results indicated that both peptides have a strong tendency to attain α-helical character, preferably from their central residues extending towards their C-terminals12-23, whereas their N-terminal residues stay either in β-turn or extended forms. However, the extent of helicity was comparatively lower in Brevinin 1-Ya, irrespective of its starting structure in the simulations, and like other factors such as cationicity and hydrophobicity, could be related to the biological activity profiles of these peptides.
Keywords: Brevinin, cationicity, anti-microbial peptides, MD, AMBER.
1. Introduction
The precise characterization of peptides is an essential condition especially when the peptides under investigations are biologically and medicinally significant and could play very important role in the design of novel potent peptidomimetics with lesser side effects. Cationic antimicrobial peptides (CAMPs) belong to this category of peptides with impressive broad-spectrum anti-fungal and antibacterial activities.1,2 Their remarkable cell-penetrating ability accounts for their cytolytic action in the mammalian cells. Thus far, these peptides have been found to exhibit very little or no resistance effects,3-5 making them excellent targets for the development of new potent anti-microbial and anti-cancer agents.
Despite having great medicinal significance, the exact mechanism responsible for the antimicrobial action of CAMPS is not clearly understood. The mostly accepted mode of action of CAMPs involves the increased membrane permeability after their interactions with them leading to the leakage of cytoplasmic components and ultimate death of cells.6-9 The positively charged ends of the CAMPs are primarily responsible for their strong interactions and bindings with the negatively charged bacterial membranes. Several studies have suggested that the increased positive charge on the peptide increases its anti-microbial action by promoting its interaction with the negatively charged bacterial membrane.10 Other factors that are considered equally important for the antimicrobial activity of CAMPS include peptide hydrophobicity3,11 and their amphipathic nature that segregates basic and hydrophobic residues12-13. Several reports have indicated the role of peptide self-association in their anti-microbial activity under the influence of membrane although no such effect was observed in the aqueous environment.12,14
Amphibians are important source of a variety of CAMPs including aureins, brevinins, maculatins and magainins.15 They are secreted by amphibian skin along with several other defensive organic compounds consisting of amines and alkaloids when these animals are exposed to the external stimuli.16 The brevinin-1 family peptides, consisting mostly of 24 amino acid (AA) residues, are isolated from the skin secretions of the leopard frogs Lithobates blairi and the Lithobates yavapaiensis, and have shown greater potency against Gram-positive bacteria.15,17 Moreover, these peptides exhibit cytolytic activity against HepG2 human hepatoma-derived cells although their anti-cancer potential has been limited by their activity against human erythrocytes. With regard to the structure-activity relationship (SAR) of the brevinin family peptides, it has been observed that replacement of Lysine AA residue at position 11 (Brevinin-1Yb) with Asparagine (Brevinin-1Ya) decreases its anti-microbial activity two fold.17 The haemolytic activity of Brevinin-1Yb also decreases to a certain extent by substituting Lys11 with Asn11. Similarly, the antibacterial activity of Brevinin-1Yc reduces significantly when the proline at position 14 is replaced with glutamic acid.17 In spite of possessing great medicinal significance, the three-dimensional (3D) structures of most of the pep-tides of the brevinin family are not fully known on the basis of available spectroscopic evidences. The circular dichroism (CD) experiments conducted on peptide Brevinin-1Ye (obtained from frog Rana esculenta) indicated the role of sodium dodecyl sulfate (SDS) concentration in the helical content of the peptide.18 In the absence of SDS the peptide did not exhibit any characteristic secondary structure feature. However, no specific information regarding the residues participating in the helicity could be ascertained from these studies.
Nowadays, X-ray and nuclear magnetic resonance (NMR) are the most popular experimental techniques used for the structure determination of peptides and other biological molecules. However, the complications associated with synthesis, isolation or crystallization of several peptides sometimes limit the use of these spectroscopic techniques in the characterization process. Moreover, the greater flexibility of the peptides in solution also make these techniques inefficient to furnish all the structural details necessary to fully understand the conformational profile of these molecules. The available computational methodologies, on other hand, have recently been proven quite useful to overcome these problems, and are extensively used to understand the conformational propensities of several biologically active peptides and proteins.
In the present study, molecular dynamics (MD) simulations have been performed on two 24 AA residue peptides, Brevinin-1Ya (Fig. 1) and Brevinin-1Yb (Fig. 1).
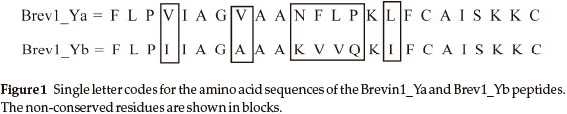
The primary goal of this work was to get a deeper understanding of the 3D structural features of these peptides. Accordingly, two MD simulations, Brev1_Ya(E) and Brev1_Yb (E),were initially carried out for 200 ns with their extended conformations as starting structures under implicit solvent conditions. The results obtained from these simulations were further compared with two additional simulations, Brev1_Ya(H) and Brev1_Yb(H) performed using the helical conformations of both peptides under similar conditions as used for the extended ones. The analysis of all trajectories was performed using the CLASICO program19 and different scripts embedded in the AMBER 9 program20.
2. Computational Methodology
All MD simulations were performed within the framework of molecular mechanics, using the all-atom parm99SB force field parameters from AMBER 9. 020. Both the extended and ~-helical conformations of the studied peptides were prepared using the tleap module of AMBER 9. Before performing simulations, the initial structures of all peptides were geometrically minimized in AMBER 9 using 1000 steps of steepest gradient, followed by a subsequent minimization using the conjugate gradient algorithm until a convergence of the gradient norm was lower than 0. 001 kcal mol-1 Å-1. Subsequently, the minimized structures were heated for 100 ps by gradually increasing the temperature from 0 to 300 K. MD trajectories were conducted using the Onufriev, Bashford and Case (OBC) implicit water model21 based on the generalized Born (GB) approximation at constant temperature (300 K) using the Langevin algorithm22 as temperature regulator. The selection of GB-OBC model in combination with FF99SB was based on its capability to accurately reproduce experimental results along with its faster sampling efficiency in the absence of solvent molecules with minimum computation power and time23-25. The simulations were performed without any cut-off. The SHAKE algorithm26 was used to constrain the bonds involving hydrogen atoms, and an integration time-step of 2 fs was employed. The internal dielectric constant for the peptide was set to 1, while an external dielectric constant of 80 corresponding to water was employed. The conformations sampled in the MD simulations were clustered using the Kleiweg's hierarchical clustering method27 to obtain further insight into the structural classification based on the secondary structures, as implemented in the CLASICO program.19
3. Results and Discussion
The structural fluctuations in the conformations of all trajectories were roughly monitored by plotting their root mean square deviation (RMSD) relative to the initial structures considering the backbone as well as all atoms, and are pictorially represented in Fig. 2a and Fig. 2b, respectively. As expected, both peptides (Brev1_Ya and Brev1_Yb) experienced larger oscillations in the trajectories starting from extended conformations than the helical ones.
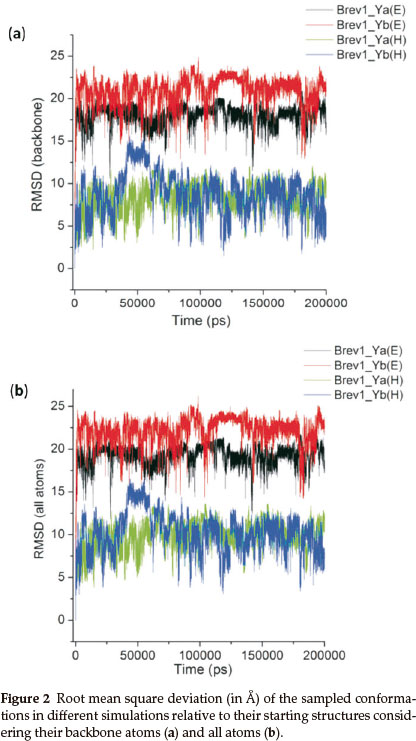
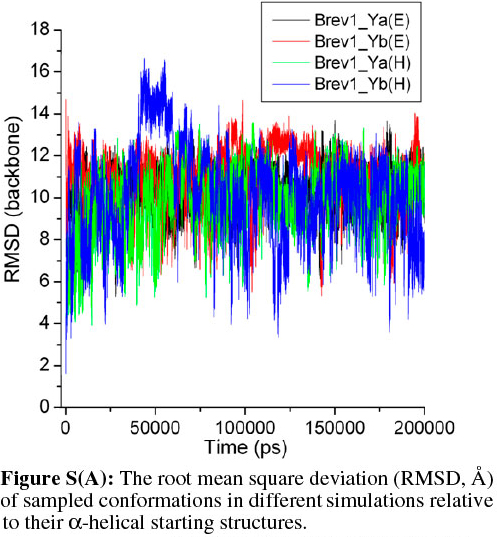
The average fluctuations around 19Å and 22Å in the Brev1_Ya(E) and Brev1_Yb(E) trajectories (Fig. 2a), respectively, clearly indicated that both peptides were sufficiently folded after few nanoseconds of the simulations, and were probably in a rapid equilibrium between the folded and unfolded structures during the whole sampling process. The considerable RMSDs (~8Å, Fig. 2a) observed in the trajectories Brev1_Ya(H) and Brev1_Yb(H), on other hand, suggested the significant loss of helicity in the sampled structures of both the peptides. The RMSD in case of all atoms (Fig. 2b) was slightly higher than the backbone atoms (Fig. 2a) indicating larger movements in the side chains of peptide during the simulation process in each trajectory. Also, the fluctuations in the atoms of Brev1_Yb were slightly higher than Brev1_Ya irrespective of the starting structures used (Fig. 2a,b). The RMSD of each simulation was further calculated considering α-helical conformation as a starting structure (Fig. S(A) in online supplement). Again, the RMSD fluctuations in the simulations starting from extended conformations were slightly higher than those starting from α-helical ones. However, the average RMSD fluctuation of each simulation around 10 Å after 100 ns suggested their convergence to comparable folded structures.
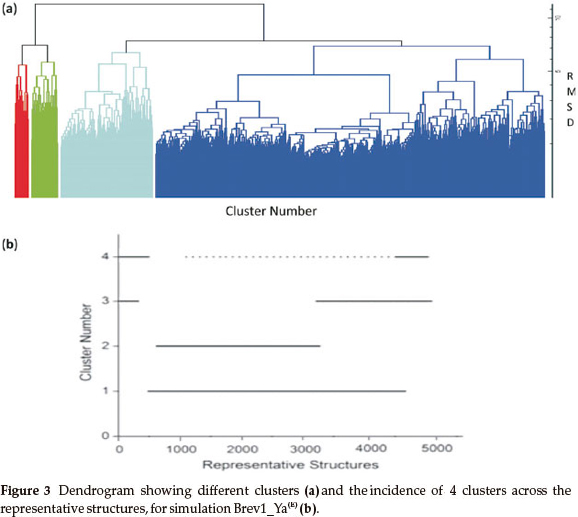
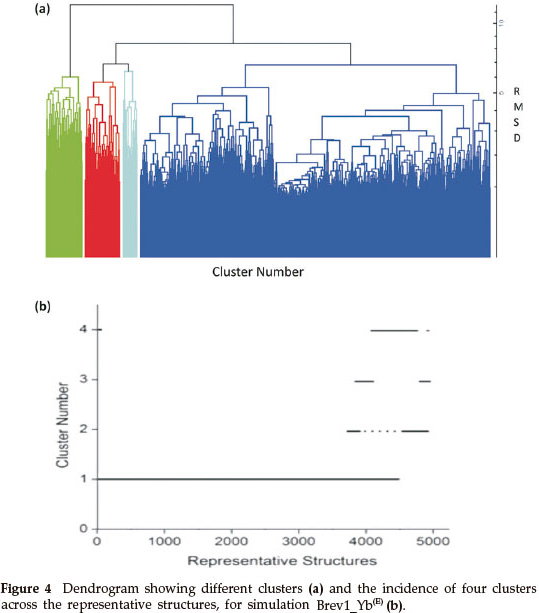
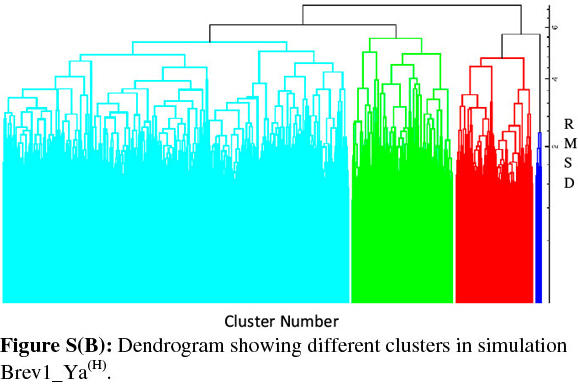
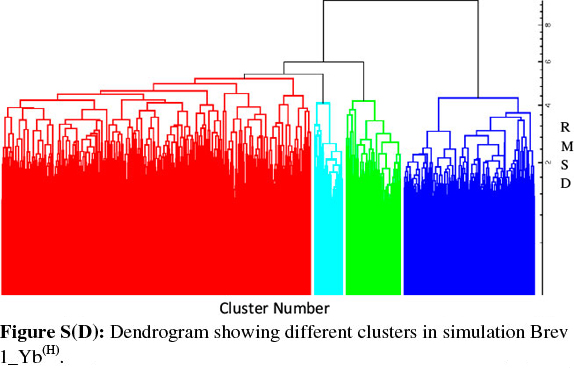
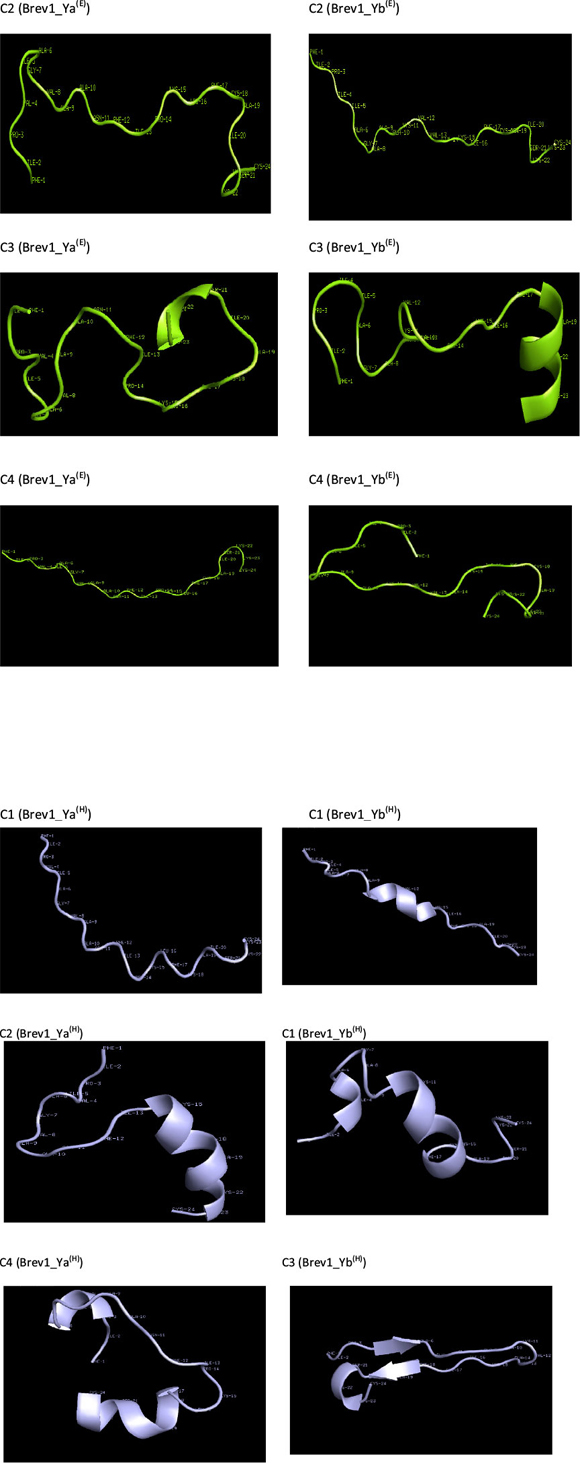
Consideringa large conformational variety in each simulation, it was thought worthwhile to group the sampled conformations into different clusters based on their structural similarity. For this purpose, the cluster analysis was performed on each MD trajectory using Kleiweg clustering algorithm.27 The 5000 conformations from each trajectory were chosen as representative structures (RS) by considering each snapshot at the interval of 40 ps. The dendrograms generated for simulations Brev1_Ya(E) and Brev1_Yb(E) are pictorially depicted in Figs 3a and 4a, respectively, while those for Brev1_Ya(H) and Brev1_Yb(H) are provided in the supplementary data (Figs S(B), S(D), respectively). The detailed results of cluster analysis are summarized in Table 1. A line drawn horizontally across the dendrogram enables us to identify the number and corresponding members of each of clusters at any particular stage. In the present cluster analysis, four major clusters have been identified (shown in different colours) irrespective of the simulation performed. A careful in- spection of Table 1 revealed C1 as the most predominant cluster in both Brev1_Ya(E) and Brev1_Yb(E) simulations containing 80% and 75% of the total structures, respectively. The second most populated cluster (C2, C4 in Brev1_Ya(E) and C3 in Brev1_Yb(E)) acquired 8% and 18% structures respectively.
The remaining clusters in both simulations (Brev1_Ya(E) and Brev1_Yb(E) contained lower percentages (less than 6%, Table 1) of the conformations. Although, the clusters C3 (66%) and C4 (60%) were predominant (Table 1) in Brev1_Ya(H) and Brev1_Yb(H), respectively, C2 and C1 also contributed 19% and 25% structures to their individual trajectories, respectively.
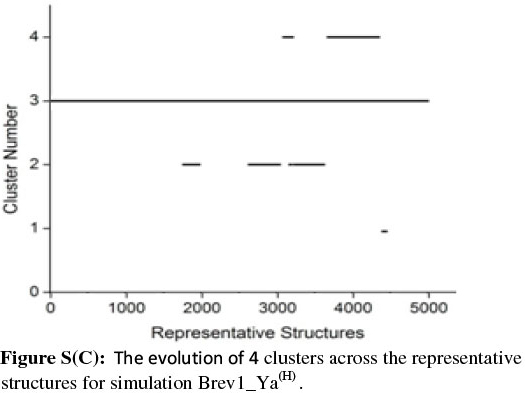
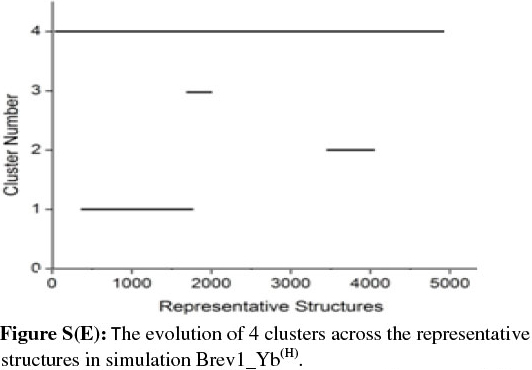
In order to get a clearer picture of the distribution of structures in each cluster, the evolution of four clusters in each simulation was plotted against the selected RS. Figs 3b and 4b represent this analysis for simulations Brev1_Ya(E) and Brev1_Yb(E), respectively, while those corresponding to simulations Brev1_Ya(H) (Fig. S(C)) and Brev1_Yb(H) (Fig. S(E)) are provided in the supplementary data.
It is clear from Figs 3b and 4b that clusters C1 in both Brev1_Ya(E) and Brev1_Yb(E) start acquiring structures immediately after the initiation of the simulations and keep adding them throughout the progress of the trajectories. Other clusters (C2, C3, C4, Figs 3b, 4b), on the other hand, acquire the conformations randomly and preferably between segments 3000 and 4500 of their respective simulations. Similarly, the major clusters C3 (Fig. S(C)) and C4 (Fig. S(E)) of Brev1_Ya(H) and Brev1_Yb(H), respectively, start growing from the beginning of the simulations and keep enriching with the new conformations throughout the sampling process. Other small clusters in both simulations again acquire new conformations in an irregular fashion in different regions of the simulations as depicted in Figs S(C) and S(E) in the online supplement.
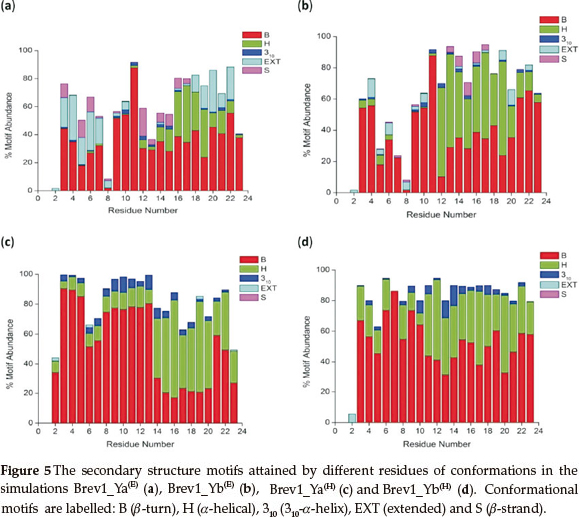
Since the main objective of cluster analysis was to get a better understanding of the structural motifs of both Brev1 peptides, further conformational analysis was focused only on the largest clusters of each trajectory. Accordingly, a qualitative analysis of the secondary motifs was performed on the conformations of each largest cluster using the CLASICO program19. This computer program translates each snapshot into a string of letters and subsequently computes for each residue its backbone torsion angles, and assigns a letter28 to it following the Zimmerman partition29 of the Ramachandran map. Thereafter, using certain rules and a three-letter window code, the secondary motif is assigned to each residue of the peptide. Histograms showing statistics of the secondary motifs per residue for each major cluster are shown in Fig. 5a-d. Specifically, the conformational motifs adopted by cluster 1 in Brev1_Ya(E) shows the predominance of β-turns in their residues (Fig. 5a). Some structures also exhibited a characteristic α-helical region flanked by residues 12-23 with a higher propensity between residues 16 and 19 (25-30%). To some extentβ-strands (3-9,12-17) and 310-helices (9-16, 19-23) were also observed in this cluster (Fig. 5a). Although, the α-helical profile exhibited by cluster 1 in Brev1_Yb(E) was quite similar to that of Brev1_Ya(E), the extent of helicity was quite high in the former especially between residues 12 and 21) as depicted in Fig. 5b. Moreover, the β-strands and 310-helices, although in lower percentages, were also adopted by some central residues of this peptide (Fig. 5b). In Brev1_Ya(H), cluster 3 acquired α-helical character almost in all residues (2-23) with a strong propensity between residues 14 and 23 and a lower propensity between residues 2 and 13 (Fig. 5c). The remaining structures of this cluster adopted predominantly β-turns. Cluster 4 of Brev1_Yb(H) also exhibited an α-helical region supported by residues 2-23 with a strong propensity between residues 11 and 23 (Fig. 5d). Interestingly, the extent of 310-helicity was comparatively higher in the simulations starting from the α-helical structures (Fig. 5a-d), and was predominantly supported by residues 9-18. Based on secondary structure analysis, it is believed that both peptides have a strong tendency to adopt α-helicity in their structures preferably from their central region expanding towards the C-terminal end. Moreover, the α-helical content was comparatively higher in Brev1_Yb than the Brev1_Ya in each simulation, especially in the proximity of its key residue (Lys11), reported to play a significant role in the activity profile of these peptides. The presence of some α-helical content (N-terminal residues) in simulations starting from helical conformations (Brev1_Ya(H) and Brev1_ Yb(H)) probably indicates the inherent tendency of such peptides to stay in helical form, suggesting the requirement of longer MD simulations or more efficient sampling techniques for these peptides to adopt their accurate native structures. However, the higher percentage of extended conformations in most of the sampled conformations in each simulation further suggested the lower conformational energy barrier between the folded and unfolded structures.
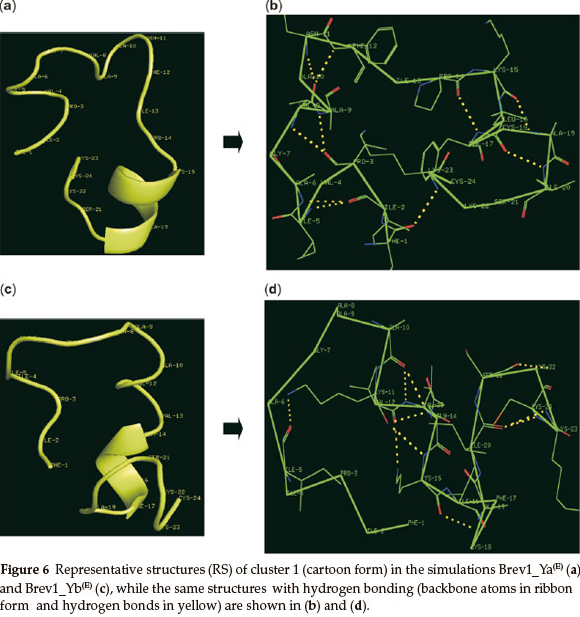
The RS of cluster 1 in Brev1_Ya(E) and Brev1_Yb(E), are diagram-matically represented in the Figs 6a and 6c, respectively. A visual inspection of Fig. 6a revealed a well defined α-helical region between residues 15 and 20. In addition, two β-turns between residues 2-5 and 16-19, and three reverse turns (RT) between residues 3-8,3-9, and 1-23, were also characterized in this RS on the basis of intramolecular hydrogen bonds (Fig. 6b). The RS of cluster 1 in Brev1_Yb(E) adopted a well defined α-helical region between residues 20 and 24 (Fig. 6c). Another distorted α-helical region between residues 10 and 17 was supported by intramolecular hydrogen bonds between residues 10-14, 10-13, and 11-14 (Fig. 6d). A characteristic loop between residues 11 and 5 and two additional β-turns between residues 15-18 and 20-23 were also observed in the same RS (Fig. 6d).
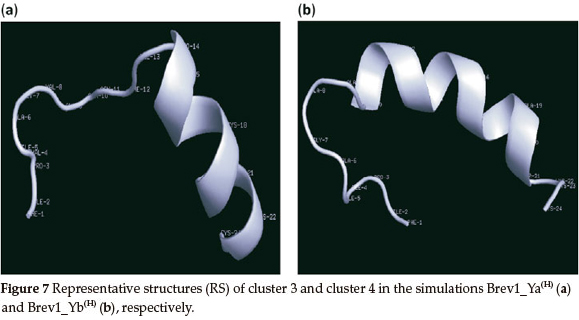
The RS of major clusters in simulations Brev1_Ya(E) and Brev1_Yb(E) exhibited more extended α-helical regions flanked by residues 14-23 (Fig. 7a) and 10-21 (Fig. 7b), respectively. The RS of remaining clusters of all simulations are provided in the supplementary section while there secondary features are summarized in Table 1.
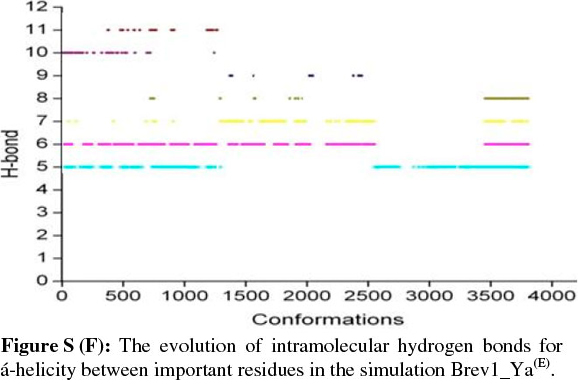
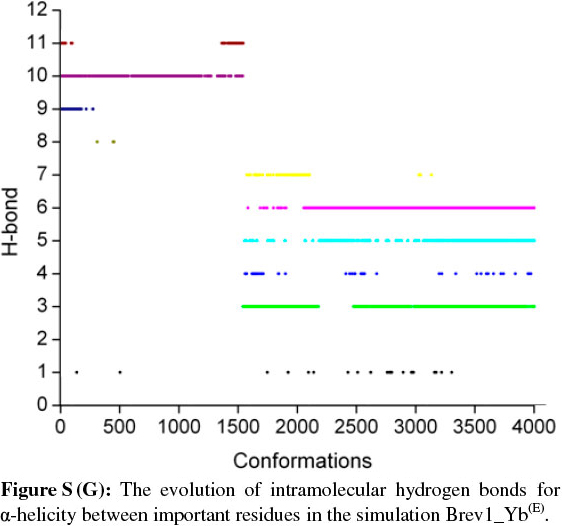
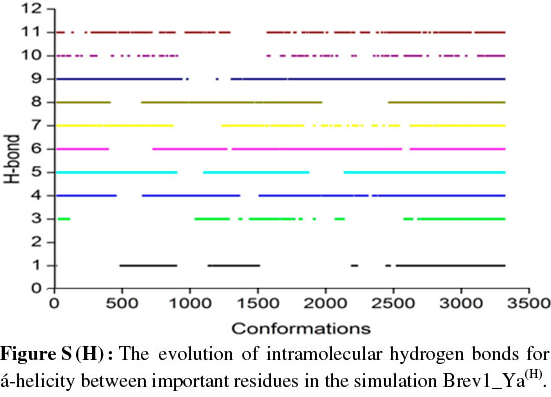
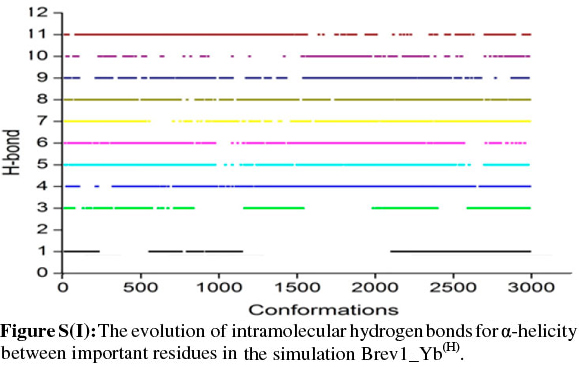
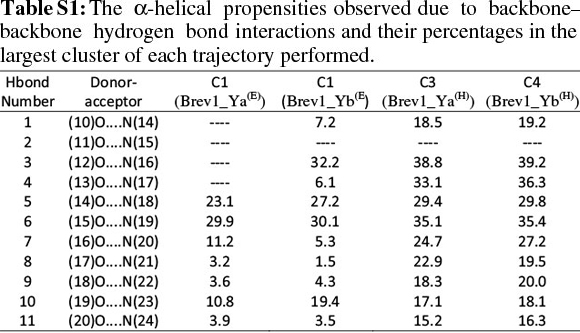
The observed α-helical propensities of the structures in the major clusters were further substantiated with an additional hydrogen bond analysis. Figs S(F-I) represents the formation/ destruction of the H-bonds across the selected RS of the largest clusters in simulations Brev1_Ya(E), Brev1_Yb(E), Brev1_Ya(H) and Brev1_Yb(H), respectively while the different results such as type of interactions, the percentage existence and the secondary features are listed in Table S1 (in online supplement). The hydrogen bonds showing percentage existence more than 1% are only considered. The geometrical criterion used involves the donor (A)-acceptor (B) distance <3.0 Å (sum of Van der Waals radii) and the angle AHB >120°, where A is donor (i.e. N-H) and B the acceptor (i.e. O=C). A closer inspection of Fig. S(F) revealed that the conformations of Brev1_Ya in cluster 1 adopted α-helicity between its central and the C-terminus residues although disrupted by some intervening residues. The extent of α-helicity was lower in the C-terminal residues, and was predominant in the central residues 14-19 (H-bond no. 5-6) of structures falling between the numbers 20 and 1300 and 3300 and 3800. The percentage of α-helicity was comparatively higher in the Brev1_Yb conformations (between 1500 and 3900) showing its predominance between residues 12 and 19 (H-bond nos 3-6) as depicted in Fig. S(G). The hydrogen bond supported by residues 19-23 (H-bond 10, Fig. S(G)) was mostly acquired by structures in the range between 10 and 1500. Although, the α-helical propensities exhibited by the Brev1_Ya and Brev1_Yb were quite similar in the simulations starting from the helical conformations (Figs S(H-I)), the percentage of helicity was again greater in Brev1_Yb(H), particularly between residues 12 and 20 (Table S1). The α-helical region between residues 10 and 24 was randomly adopted by most the conformations throughout the progress of the simulation (Figs S(H-I)). Interestingly, no hydrogen bond between residues 11 and 15 was observed in the major clusters of any simulations (Figs S(F-I)). Another common feature observed in these simulations was the lower content of helicity in the C-terminal residues than the central ones, and was in accordance with the results obtained from the secondary structure analysis as shown in Figs 5a-d.
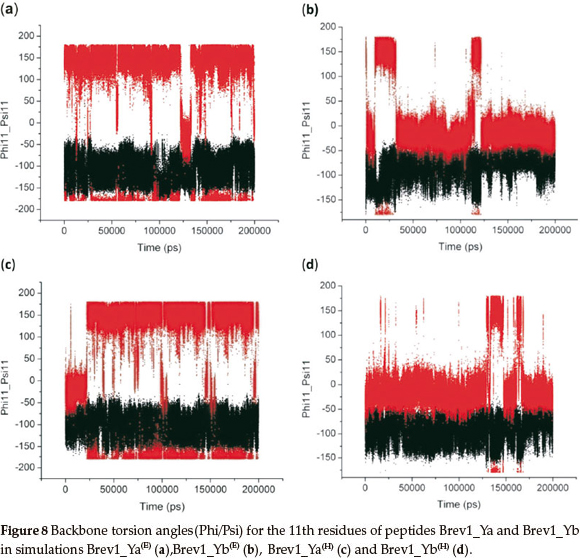
Finally, keeping in view the significance of residue 11 in the biological activity of these peptides, the conformational behaviour of this AA was monitored by plotting its dihedral angles (Phi11/Psi11) as a function of time in all simulations, and is picto-rially represented in Fig. 8a-d. A similarity in the conformational preferences was observed within the simulations of the same peptide irrespective of the initial structure used. Fig. 8a indicates that Asn11 in Brev1_Ya(E) exhibited significant fluctuations (-180 to 180) in its Phi angle (C-N-Cα-C), while its Psi angle (N-Cα-C-N) remains relatively fixed (-100), adopting the β-sheet conformation. The transformation of initial right-handed α-helical (αr) configuration of Asn11 into β-sheet after ~20 ns of Brev1_Ya(H) (Fig. 8c) also further supported its stability in this form. The initial conformation of Lys11 residue in Brev1_Yb, on other hand, remains relatively fixed (Phi11 = -10 and Psi11 = -95) in its both trajectories (Fig. 8b,d) suggesting αR as its preferred conformation. Hence, it is believed that the substitution of Lys11 in Brev1_Yb with Asn11 probably decreases the helical tendencies of this peptide, and seems to be one of the contributing factors for its decreased activity after the substitution. The strong α-helical favouring nature of Lysine over Asparagine residue30 also supports the higher α-helical propensities of Brev-1Yb observed over its structural analogue Brev-1Ya. The relationship between activity and α-helicity of peptides considered in the present study, was based on reported articles where increased biological activity of the peptides was found to be proportional to their α-helical content. For instance, the antifreeze activity of winter flounder increases significantly with the helical content in its structure.31 Similarly, the increased inhibiting tendency of T-1249 relative to that of T-20 (anti-HIV drug) is believed to be related to its higher α-helical content.32
4. Conclusions
Considering the great medicinal and biological significance of brevinin 1 family peptides, four MD simulations were performed on two brevinin 1 peptides (Brev1_Ya and Brev1_Yb) aimed at understanding the conformational preferences of these peptides. Analysis of the results indicated that both peptides attain a well defined α-helical region flanked by their central and C-terminal residues (although in different percentages) in all simulations. However, the extent of helicity was lower in Brev1_Ya than Brev1_Yb in all simulations irrespective of the starting geometry used, and could be playing some role in the decreased biological activity of this peptide after substitution of Lys11 with Asn11. The substantial transformation of α-helical configuration of Asn11 into β-sheet in simulation Brev1_Ya(H), and its retention in Brev1_Ya(E) also supported the observed lower helical propensities of Brev1_Ya, and its some role in their biological activity profiles. It is worthwhile mentioning that the folded structures sampled in all trajectories were not stable enough, and were in a rapid equilibrium with the unfolded/extended conformations throughout the simulation process suggesting a low conformational energy barrier between them. The observed structural propensities of brevinin 1 peptides in the present study would definitely increase our understanding about their bioactive conformations and activity profiles, and could open channels for the design of new peptidomimetics with similar or better biological functions.
Acknowledgements
Financial support from the Durban University of Technology (DUT) and the National Research Foundation (NRF) is gratefully acknowledged. I am highly thankful to Professor Krishna Bisetty and our Spanish collaborators (Professor Juan J. Perez, Dr. Francesc Corcho and Dr. Alex Rodriguez) for training and regular research support.
References
1 R.E.W. Hancock, Lancet Infect. Dis., 2001, 1, 156-164. [ Links ]
2 M. Zasloff, Nature, 2002, 415, 389-395. [ Links ]
3 R.E.W. Hancock, Lancet Infect. Dis., 2005, 5, 209-218. [ Links ]
4 D.A. Devine and R.E.W. Hancock, Curr. Pharm. Des., 2002, 8, 703-714. [ Links ]
5 A. Peschel and H.G. Sahl, Nat. Rev. Microbiol., 2006, 4, 529-536. [ Links ]
6 D.I. Chan, E.J. Prenner and H.J. Vogel, Biochim. Biophys. Acta, 2006, 1758, 1184-1202. [ Links ]
7 H. Jenssen, P. Hamill and R.E. Hancock, Clin. Microbiol. Rev., 2006, 19, 491-511. [ Links ]
8 K.A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238-250. [ Links ]
9 Y. Shai, Biochim. Biophys. Acta, 1999, 1462, 55-70. [ Links ]
10 J.M. Conlon, N. Al-Ghaferi, B. Abraham and J. Leprince, Methods, 2007, 42, 349-357. [ Links ]
11 Y. Chen, M.T. Guarnieri, A.I. Vasil., M.L. Vasil, C.T. Mant and R.S. Hodges, Antimicrobial Agents and Chemotherapy, 2007, 51,1398-1406. [ Links ]
12 Y. Chen, C.T. Mani, S.W. Farmer, R.E. Hancock, M.L. Vasil and R.S. Hodges, J. Biol. Chem., 2005, 280, 12316-12329. [ Links ]
13 R.M. Epand and H.J. Vogel, Biochim. Biophys. Acta, 1999, 1462, 11-28. [ Links ]
14 D.L. Lee, C.T. Mant and R.S. Hodges, J. Biol. Chem., 2003, 278, 22918-22927. [ Links ]
15 E.F. Haney, H.N. Hunter, K. Matsuzaki and H.J. Vogel, Biochim. Biophys. Acta, 2009, 1788, 1639-1655. [ Links ]
16 T.L. Pukala, J.H. Bowie, V.M. Maselli, I.M. Musgrave and M.J. Tyler, Nat. Prod. Rep., 2006, 23, 368-393. [ Links ]
17 J. M. Conlon, E. Ahmed, L. Coquet, T. Jouenne, J. Leprince, H. Vaudry and J.D. King, Lithobates blairi and Lithobates yavapaiensis, Toxicon, 2009, 53, 699-705. [ Links ]
18 M. Kwon, S. Hong and K. Lee, Biochim. Biophys. Acta, 1998, 1387, 239-248. [ Links ]
19 LaFargaCPL: CLASTERIT: Project Info. Online at: http://devel. cpl. upc. edu/clasterit. [ Links ]
20 D.A. Case, T.A. Darden, T.E. Cheatham III, C.L. Simmerling J. Wang, R.E. Duke, R. Luo, K.M. Merz, D.A. Pearlman, M. Crowley, R.C. Walker, W. Zhang, B. Wang, S. Hayik, A. Roitberg, G. Seabra, K.F. Wong, F. Paesani, X. Wu, S. Brozell, V. Tsui, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, P. Beroza, D.H. Mathews, C. Schafmeister, W.S. Ross and P.A. Kollman, AMBER 9, University of California, San Francisco, 2006. [ Links ]
21 A. Onufriev, D. Bashford and D.A. Case, Proteins: Struct. Funct. Bioinf., 2004, 55, 383-394. [ Links ]
22 X. Wu and B.R. Brooks, Chem. Phys. Lett., 2003, 381, 512-518. [ Links ]
23 Y. Shang, H. Nguyen, L. Wickstrom, A. Okur and C. Simmerling, J. Mol. Graph. Model., 2011, 29, 676-684. [ Links ]
24 M.S. Shell, R. Ritterson and K.A. Dill, J. Phys. Chem. B., 2008, 112, 6878-6886. [ Links ]
25 J. Mongan, C. Simmerling, J.A. McCammon, D.A. Case and A. Onufriev, Generalized Born model with a simple, robust molecular volume correction, J. Chem. Theory Comput. 2007, 3, pp. 156-169. [ Links ]
26 J-P. Ryckaert, G. Ciccotti and H.J.C. Berendsen, J. Comp. Phys., 1977, 23, 327-341. [ Links ]
27 P. Kleiweg, J. Nerbonne and L. Bosveld, 2004, Geographical projection of cluster composites, in Diagrammatic Diagrams 2004, (A. Blackwell, K. Marriott and A. Shimojima, eds),Lecture Notes in Computer Science, Springer, New York, 392-394. [ Links ]
28 F. Corcho,J. Canto andJ.J. Perez, J. Comp. Chem, 2004, 25, 1937-1952. [ Links ]
29 S. S. Zimmerman, M.S. Pottle, G. Nemethy and H.A. Scheraga, Macromol., 1977, 10, 1-9. [ Links ]
30 M. Levitt, Biochem.,1978,17, 4277-4285. [ Links ]
31 A. Chakrabartty and C.L. Hew, Eur. J. Biochem, 1991, 202, 1057-1063. [ Links ]
32 A.M.T.M.D. Canto, A.J.P. Carvalho, J.P.R. Ramalho and L.M.S. Loura, J. Pept. Sci., 2008, 14, 442-447. [ Links ]
Received 31 July 2012
Accepted 21 February 2013
* E-mail: parveshdurban@gmail.com / parveshs@dut.ac.za
Supplementary Data


{kind=link}