










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
RESEARCH ARTICLE
Terpyridyl complexes as antimalarial agents
Jason N. ParaskevopoulosI; Peter J. SmithIII; Heinrich C. HoppeIV; Deepak ChopraV; Thavendran GovenderII; H.G. KrugerI; Glenn E.M. MaguireI, *
ISchool of Physics and Chemistry, University of KwaZulu-Natal, Westville Campus, Durban, 4000, South Africa
IISchool of Pharmacy and Pharmacology, University of KwaZulu-Natal, Westville Campus, Durban, 4000, South Africa
IIIDivision of Pharmacology, University of Cape Town, K45, OMB, Groote Schuur Hospital, Observatory, 7925, South Africa
IVDepartment of Biochemistry, Microbiology and Biotechnology, Rhodes University, P.O. Box 94, Grahamstown, 6140, South Africa
VDepartment of Chemistry, Indian Institute of Science Education and Research, Bhopal 462 023, India
ABSTRACT
A number of transition metals and their terpyridyl complexes have been evaluated for antimalarial activity on the strain 3D7. The metals, ligands and complexes were each in turn investigated for their efficacy. All activities were in the sub-micromolar range (0.1-1 µM). Their modes of action were compared with that of chloroquine to discover whether or not they were capable of inhibiting haemozoin formation. The data indicate that efficacy could be a result of several mechanisms and that speciation of the metal complex and the manner in which the agents are added to the parasitic broth have a profound effect on the activity of the agents. We believe that our study offers a template by which other researchers should approach their experiments using transition metal complex agents.
Keywords: Antimalarial agents, terpyridyl and transition metals.
1. Introduction
As of 2004 malaria was endemic in 107 countries with approximately 3.2billion people exposed to infection. It is estimated that between 300 and 500 million clinical cases occur per year, caused primarily by Plasmodium falciparum and Plasmodium vivax. Plasmodium falciparum is directly responsible for more than one million deaths (mostly African children under the age of five) annually, and may contribute to even more deaths in combination with other illnesses. More than 80 % of these deaths take place in sub-Saharan Africa. The mortality effect of malaria in Africa is amplified by the high rate of HIV/AIDS infections as well as the inability of public health systems to deal with such high infection rates.1
The transportation and accumulation of metal ions into the malaria parasite is one aspect of the parasite which is of particular interest as a potential drug target.2 While a significant amount of work has been performed on the transport and uptake of organic solutes, there has in contrast been very little research on the parasites preference for metals, especially for the transition metals of copper, iron, gold and platinum. For copper and iron there is some, though little, research on the parasites requirement for these metals. Due to the trace amounts of gold and platinum in the human body it is not surprising that there is no report of the infected red blood cells' (iRBCs) ability to transport these metals.
It has been shown that during the parasite's life cycle it has no need to accumulate copper, since the amounts present within the RBC prior to invasion are sufficient to support the parasite.3 It was even reported that the parasite demonstrates pathways which in fact remove copper from the infected erythrocyte, leaving the infected blood cell with approximately 66 % of the average amount found in a normal erythrocyte.3 Thus it appears that the parasite has an efficient copper efflux system to remove the toxic metal which is freed during the degradation of erythrocyte proteins.4 In the case of iron there has been evidence to both suggest and refute that the parasite obtains this element from the serum, from the erythrocyte as free iron or from degraded haemoglobin. Reports in favour of transport from the serum, show evidence for transferrin-independent iron translocation5 and the resistance to malaria that people suffering from anaemia have demonstrated.6,7 Arguments against the serum as a source of iron stem from the lack of transferrin receptors on iRBC membranes,8 that iron-supplemented diets had no effect on the course of malaria in infected patients9 and that there was a minimal increase in the uptake of radiolabelled iron in iRBCs when compared with normal RBCs.10 In one study iron sequestration agents were preloaded into erythrocytes and shown to have little or no effect on the invading species activity suggesting that free erythrocytic iron may not be required by the parasite.11 Haemoglobin degradation might seem an obvious source of this metal;12 it has been demonstrated by Egan et al. that 95 % of the parasite-digested haem iron can be found in haemozoin. The parasites source of iron is thus disputed. Scheibel et al. did, however, also prove that transport of this metal ion, and presumably others, such as copper, gold and platinum, can be mediated by ligands such as 8-hydroxyquinoline (uptake of serum iron increased from 9 % to 50 % in the presence of 8-hydroxyquinoline).9
Surprisingly, the general approach to the application of metal 'complexes' in malarial experiments is not uniform. Not only have researchers not tested the background activity of the metals but also on many occasions the metal ion and ligand were added as a 'cocktail' solution with the expectation that a complex will form. The exact ratio of metal to ligand and thereby the active species is thus indiscernible.
What little research there is on the transport of metal ions (perhaps with the exception of sodium and calcium) into the infected erythrocyte, suggests that in general transportation of metals into the parasite is likely to be mediated by organic ligands.
In light of the paucity of literature studies on their antimalarial activity, it was decided to examine the activity of these metals using a ligand whereby the corresponding metal complexes are water soluble under biological conditions. Only on a single occasion have some terpyridyl (1) derivative complexes been tested for their antimalarial activities in a patent released by Lowe.13 However, neither the parent ligand nor its complexes with transition metals, Au, Cu, Fe, Pt and Pd, have been tested for antimalarial activity. In this paper we report the evaluation of the activity of such systems. In particular, we employ control experiments that evaluate whether the assumption of active 'complex' idea is a sensible one.
2. Results
2.1. Antimalarial Testing
As part of examining the roles of metals in this study it was important to first determine the background activities of the transition metals of interest. The ions investigated in this study included Cu2+14-16, Fe3+9.14.15, Au3+ and Pt2+ (with terpyridyl).17-19 The toxic effect of each of the metals was determined using their chloride salts. These results can be seen in Table 1.
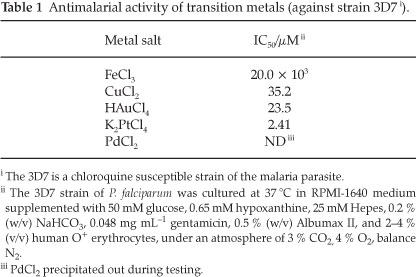
With the exception of the platinum salt, the toxicities of the metals are quite low, and as one might expect in a biological system, the toxicity increases in the order Fe3+ <Cu2+ <Au3+ < Pt2+. With these values established it was decided that the metals would have minimal toxic effect at concentrations of 0.5 µM (as free metals in the growth medium).
The second part of this study was aimed at determining whether or not having such a significant metal concentration in the growth medium would potentiate the activity of the ligand. In these experiments it was anticipated that the terpyridyl ligand would form a 1:1 complex with the specific metals. Terpyridyl, alone, showed an IC50 of 1.0 µΜ against the 3D7 strain of malaria. The activities of the different metal background solutions are reported in Table 2.

A notable result is that of terpyridyl with free iron. The activity is much weaker than that observed for the ligand alone (1.0 µM, a more than ten-fold reduction in activity) and therefore suggests that iron is antagonistic towards the activity of the ligand. This is likely due to complexation of free iron by terpyridyl which either prevents its mode of action or alters uptake of the ligand or its transportation into the parasite. Since the other metals do not inhibit the activity to such an extent it seems logical to suggest that the complex formed in situ did in fact prevent the antimalarial mode of action by reducing its transportation into the parasite. This is possibly due to the relatively high effective charge density associated with the terpyridyl-iron(III) complex. The ligand also shows reduced activity in the presence of free gold(III), IC50 2.3 µM, although it is not as pronounced as found in the case of iron (III). This could be due to the larger size of the gold (III) ion, leading to a lower effective charge density. Free copper and platinum, both having charges of 2+, in this study demonstrated no observable effect on the activity of terpyridyl.
Importantly these results have three potential implications. First, it suggests that the mode of action of these ligands does not involve the formation of a toxic metal complex in situ. If this were the case, one would expect a rise in activity with an increase in background metal concentration (as observed in antibacterial studies).15 Second, the ligands' activity does not depend on the sequestration of these metals away from the parasite, since the toxic effect is not reversed by the addition of these free metals. Third, this lack of reversal also suggests that the mode of action is not mediated via coordination to a metalloenzyme.20 The free metals should be as easily complexed as enzyme bound metals thus decreasing the amount of free ligand available to block the parasite-enzyme metal sites.
Next, complexes of the ligand with each of the metals were made. Single crystal X-ray structures were obtained for the copper21 and iron22 terpyridyl complexes and they are shown in Figs 1 and 2, respectively. We have also published the redetermined structure of gold(III) terpyridyl chloride.23 The anti-malarial activities of these terpyridyl complexes are reported in Table 3.



As seen in Table 3, each of the complexes were found to have a slightly better activity than terpyridyl as a free ligand (IC50 = 1.0 µM). The terpyridyl-gold(III) demonstrates the highest activity; approximately three-fold of that seen for the ligand on its own. If one views these results from the perspective of the activity of terpyridyl, it appears as if the metals do little to enhance the activity of the ligand. Furthermore it suggests that metal complexation is not essential for terpyridyl to exhibit its anti-malarial effect. In cancer studies it has predominantly been suggested that metals are required for terpyridyl to exhibit activity,18,19,24 although a few papers have shown that terpyridyl and some derivatives have similar or even higher anticancer activity than their complexes.25-27 It is often the case in literature studies that the free ligands are not tested for any intrinsic activity.
These results can also be viewed from the perspective of the metals. For each of the metals the activity has been increased significantly. A comparison of the activities of the metal salts and the terpyridyl-metal complexes is shown in Table 4.
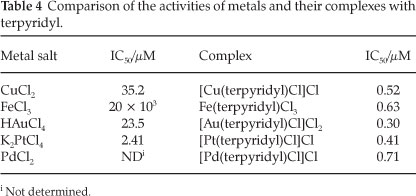
The increase in efficacy is most pronounced for iron(approximately 30 000-fold), followed by gold (approximately 78-fold), copper (approximately 68-fold) and platinum (approximately six-fold). This observation concerning the metals is quite possibly due to their enhanced transportation into the parasite upon complexation. No direct measurement of the transportation (or uptake) of the metals was performed. It has, however, been demonstrated previously that ligands are capable of facilitating the transport of metal ions into the parasite.9 Thus it could well be that the ligand allows more of the metal ions to reach their active site(s) in the parasite.
It is interesting to note that the results are open to interpretation from either the ligand perspective (which suggests only a marginal increase in activity) or from the metals' perspective (which then suggests a significant increase in activity). If the exact modes of action of the ligand, the metals and the complexes were all known, one could perhaps determine which of these aspects holds more significance. It is of course quite possible as well that they all act via totally different mechanisms. It is now a generally held belief that chloroquine exhibits its antimalarial effect by inhibiting haemozoin formation; whether it is by inhibiting an enzyme which produces haemozoin or by preventing a spontaneous reaction is debatable.28,29 In light of this, it became essential to test whether any of our agents demonstrate such ability. A method devised to establish whether potential antimalarial compounds are capable of performing this feat has been reported by Egan et al.30
2.2. β-Haematin Inhibition
In these experiments chloroquine was used as a standard. The ligand was also tested to see if it possibly had the same mechanism as chloroquine. It was suspected that this compound might demonstrate some ability to inhibit/3-haematin formation due to its planar structure. Each of the metal salts and in turn their complexes was evaluated. The results of these investigations are presented in Table 5.

As seen in Table 5, the terpyridyl alone does not, however, seem to operate via an inhibition mechanism. It was found that the metal salts of iron, gold and platinum were capable of inhibiting β-haematin formation, while that of copper did not. The same result was also observed for the metal complexes. This suggests that the ability to inhibit β-haematin formation is inherent to the metals, and not a new property of the ligand-metal complex. The activity of the free metals may well be derived from their ability to coordinate to the heme groups via the propionic functional groups and thus create a form of aggregation.
These results contrast, however, with other reported platinum pyridyl complexes. Egan et al.31 suggested that the ability of these complexes to inhibit β-haematin formation could be attributed to their self-aggregating behaviour, a property likely due to the planar nature of the ligand-metal complexes. Our results, however, imply that the β-haematin inhibiting properties of any complexes of iron, gold or platinum, may be due to the metals' inherent ability to exhibit this action, and not in fact due to the combination of ligand(s) and metal, as has been reported.
Interestingly the results here also imply that ligands which do not inhibit β-haematin can have this property conferred on them by metals such as iron, gold and platinum. It confirms the view that in general ligands can act via different mechanisms from their corresponding complexes and furthermore that ligands in the complex form may only be responsible for increasing the transport (and uptake) of these toxic metals into the parasite. The activity observed for many complexes may in fact be more a direct result of the properties of the metal centre.
It should be noted that copper did not demonstrate any ability to inhibit β-haematin formation, neither as the free metal nor in complex form and yet the terpyridyl-copper complex still shows higher activity than the free metal. Unfortunately we have no explanation at this time as to why copper and its associated complex behave in this way.
3. Discussion
This is the first apparent report of the antimalarial activity of both terpyridyl and its complexes with the metal ions of copper, iron, gold, and platinum. In order to study the relationship between the ligands and metals, several experiments were performed. The background activity of each metal was measured. Their toxicities ranged from 2.4 µM (platinum) to 20 mM (iron).With these values established, the relationship between the activities of terpyridyl and the free metal concentrations in solution was investigated. It was found that with the exception of iron, the free metal concentration had neither an antagonistic nor synergistic effect on the antimalarial activity of the ligands. High concentrations of free iron were antagonistic toward the activity of terpyridyl (ten-fold reduction of the IC50 to approximately 10 µM). The metal-ligand complexes were successfully synthesized and these complexes showed activity similar to that of the ligand alone. All activities were in the sub-micromolar range (0.1-1µM).
As a secondary study the ability of each of the compounds to inhibit S-haematin formation was investigated. While terpyridyl showed no ability to inhibit β-haematin formation, it was revealed that β-haematin inhibition was intrinsic to the metals iron, gold, platinum and palladium (results not shown) but not copper. It was also found that all of the complexes of terpyridyl, with the exception of that of copper, inhibited S-haematin formation. Thus it is apparent that the complexes of terpyridyl, and the ligand itself, might well exert their toxic effects by different mechanisms. Unusually our study demonstrates the possibility that ligands serve to enhance the activity of the metals, an idea which could be viewed as somewhat opposite to the conventional perspective that metals increase the activities of ligands.
4. Experimental
All solvents and reagents were obtained from Acros, Aldrich, Fluka, Merck or Alfa-Aesar. Unless otherwise stated, these were used without further purification. Solvents (CH3CN and DMF) were dried according to the methods in Vogel.32 All other solvents were used without drying. Thin-layer chromatography (TLC) was conducted on aluminium-backed, pre-coated silica gel plates (Merck, silica gel 60,20 cm x 20 cm). Column chromatography was performed with silica gel 60 (Merck, particle size 0.040-0.063 mm). Proton NMR spectra were recorded at 298 Kat 300 MHz on a Varian Unity Inova spectrometer; carbon NMR spectra were recorded at 75 MHz with the same instrument under the same conditions. Instances where proton and carbon NMR spectra were recorded at 400 MHz, a Varian Oxford spectrometer or a Bruker Advance spectrometer were used, at an operating temperature of 298 K. NMR spectra recorded at 600 MHz were performed under the same condition using a Bruker Ultrashield spectrometer. Chemical shifts are reported in parts per million (ppm) and refer to the respective deuterated solvents as reference, CDCl3 (1H 7.24 ppm, 13C 77.0 ppm), d6-DMSO (1H 2.49 ppm, 13C 39.5 ppm) and D2O(1H 4.79 ppm, 13C ppm referenced to CH3CN). Infrared spectra were collected at 293 Kby attenuated total reflection (ATR) using a Perkin Elmer Universal ATR Sampling Accessory attached to a Spectrum 100 FT-IR Spectrometer. Wave numbers are reported in units of cm-1. All melting points are uncorrected. Mass spectra were recorded using a Bruker micrOTOF-Q II Electron Spray Ionization (ESI) Mass Spectrometer (MS).
[Pt(terpyridyl)Cl)]Cl.2H2O33
Potassium chloroplatinate(II) (0.100 g, 0.241 mmol) was dissolved in H2O (5 mL). Solid 2,2':6',2''-terpyridine (0.056 g, 0.24 mmol) was then added to this solution followed by further H2O (2 mL). The orange solution was then stirred under reflux overnight. The reaction mixture was then cooled, any particulate matter filtered and the solvent removed under reduced pressure. The resulting solid was then dissolved in a minimum amount of H2O and the product precipitated out by the addition of HCl (conc. approx. 10 drops). The bright orange productwas filtered off and dried in a vacuum desiccator (0.042 g, 33 %).Mp >300 °C. <5H (400 MHz, DMSO-d6) 8.89 (dd, J=1.5 Hz,J=5.5 Hz, 2H), 8.64 (m, 5H), 8.51 (td, J=8.0 Hz, J=1.5 Hz, 2H), 7.95 (m, 2H). °C (100 MHz, DMSO-d6) 158.3, 154.4, 151.3, 142.7, 142.1, 129.2, 125.9,124.5. I.R. (vmax/cm-1) 3297,3031,1604,1476,1451,1401,1314, 1249, 1091, 1030, 779, 721, 593, 546, 521, 461. Calculated for PtC15H11N3Cl+m/z = 463.0289. Found: m/z = 463.0282 [M].
[Au(terpyridyl)Cl]Cl2.3H2O34
2,2':6',2''-Terpyridine (0.067g, 0.29 mmol) was added to a solution of HAuCl4.H2O (0.100 g, 0.294 mmol) in H2O (7.5 mL). This was followed by the addition of HCl (1 M, 375 µL, 0.375 mmol). The solution formed a yellow precipitate. The pH was then adjusted to between 3 and 5 by adding NaOH (1 M, 240 µL, 0.240 mmol) giving an orange-red solution with a yellow precipitate. The reaction mixture was refluxed overnight dissolving most of the precipitate. After hot filtration, the solvent was removed and the product recrystallized from H2O (0.090 g, 53 %). Mp 245-250 °C (decomp.). H (400 MHz, DMSO-d6) 8.77 (dd,J=1.5 Hz,J=5.0 Hz, 2H), 8.71 (d,J=8.0 Hz, 2H), 8.49 (d,J=8.0 Hz, 2H), 8.15 (m, 3H), 7.60 (m, 2H). δC (100 MHz, DMSO-d6) 151.8, 151.5, 146.8, 141.5, 139.7,126.0, 122.8,122.7. I.R. (vmax/cm-1) 3306,3010,1602,1476,1449,1401,1322,1249,1089,1028, 772, 761, 656, 542, 514, 441. Calculated for AuC15H11N3Cl2+m/z = 499.9996. Found: m/z = 499.9978 [M].[Cu(terpyridyl)Cl]Cl35 2,2':6',2''-Terpyridine (0.118 g, 0.506 mmol) was dissolved in methanol (20 mL) and added to a solution of CuCl2.2H2O (0.084 g, 0.493 mmol) in methanol (10 mL). The terpyridine solution was added to the copper(II) chloride solution resulting in a colour change from light to dark green, followed by the formation of a green precipitate. The solution was stirred for ten min and then left in a freezer for three days. The product, a dark green powder, was filtered off and recrystallized from water to afford dark green crystals of the paramagnetic product (0.139 g, 75 %). Mp >300 °C. I.R. (vmaxcm-1) 3338,3215,1631,1595, 1562, 1489, 1428, 1407, 1302, 776, 737, 648, 625, 437, 408. Calculated for CuC15H11N3Cl+m/z = 330.9938. Found: m/z = 330.9936 [M]-Fe(terpyridyl)Cl336 2,2':6',2''-Terpyridine (0.100 g, 0.430 mmol) was dissolved in methanol (5 mL) and added to a solution of FeCl3.6H2O (0.111 g, 0.411 mmol) in methanol (15 mL). The terpyridine solution was added to the iron(III) chloride solution, resulting in a colour change from yellow to red, followed by the formation of a yellow precipitate. The solution was stirred for one hour at room temperature and filtered. The product was recrystallized from water to afford amber crystals of the paramagnetic product (0.122g, 75 %).Mp >300 °C. I.R. (vmaxcm-1) 3075,1596,1574,1475, 1444, 1317, 1244, 1155, 1096, 1021, 780, 733, 650, 512, 415. Calculated for FeC15H11N3Cl2+m/z = 358.9679. Found: m/z = 358.9678 [M].
[Pd(terpyridyl)Cl]Cl.3H2O from PdCl237
Palladium(II) chloride was dissolved in H2O (10 mL) and HCl (32 %, 3.00 mL, 31.5 mmol) and heated to reflux. The reaction mixture became clear red and was left for a further 45 min. The warm solution was filtered and a solution of 2,2':6',2''-terpyridine (0.120 g, 0.514 mmol) in methanol (10 mL) was added dropwise to the filtrate. The newly formed peach-coloured precipitate was filtered off and the pH of the filtrate adjusted to between 4.5 and 5 using NaOH (2 M). The solution was again filtered and the filtrate left until yellow-orange crystals formed (0.045 g, 19 %).Mp >300 °C. δH (400 MHz, DMSO-d6) 8.72 (dd, J=1.5 Hz, J=5.5 Hz, 2H), 8.64 (m, 5H), 8.46 (td, J=8.0 Hz, J=1.5 Hz, 2H), 7.88 (m, 2H). °C (100 MHz, DMSO-d6) 158.0,154.6,152.0, 142.8, 142.6, 128.9, 125.4, 124.5. I.R. (vmax cm-1) 3307, 3016, 1601, 1448, 1400, 1320, 1247, 1088, 1027, 774, 719, 657, 566, 514, 440. Calculated for Pd(C15H11N3)Cl+m/z = 373.9676. Found: m/z = 373.9675 [M].
4.1. Parasite Culture
The 3D7 strain of P. falciparum was cultured at 37 °C in RPMI-1640 medium supplemented with 50 mM glucose, 0.65 mM hypoxanthine, 25 mM Hepes, 0.2 % (w/v) NaHCO3, 0.048 mg mL-1 gentamicin, 0.5 % (w/v) Albumax II, and 2-4 % (v/v) human O+ erythrocytes, under an atmosphere of 3 % CO2, 4 % O2, balance N2.
4.2. Parasite Viability Dose-Response Assays
Culture-derived parasitized erythrocytes were mixed with fresh culture medium and erythrocytes to yield a 2 % parasitemia, 2 % haematocrit suspension and distributed in microlitre plates at 100 µL/well. Serial dilutions of test drug in culture medium were prepared in quadruplicate wells in a separate plate and transferred to the parasite plate to yield a final volume of 200 µL/well. The plates were incubated at 37 °C for 48 hours and parasite viability in each well measured by the colorimetric determination of parasite lactate dehydrogenase activity. All experiments were performed in triplicate.
4.3. Inhibition of β-Haematin Formation Assays
A sample of haemin (15 mg, 0.023 mmol) was placed in a test tube with NaOH (0.1 M, 3 mL). This mixture was heated to 70 °C. To this solution was added first an acetate buffer solution (12.9 M, pH 5, 1.75 mL) followed by HCl (1 M, 0.30 mL) both pre-incubated at 70 °C. After stirring for two hours in a temperature-controlled bath, the test tube was placed on ice for five minutes. In the study by Egan et al.30 β-haematin was found to form within 30 min at 60 °C; however, here it was found that the formation of β-haematin was only complete after two hours at 70 °C. Once cooled the contents of the test tubes were transferred to vials, and centrifuged at 6000 rpm for five minutes. The supernatant was decanted, and the remaining solid was shaken with deionized water (8 mL) and centrifuged again at 6000 rpm for five minutes. The solid was washed in this way three times following which, while still wet, it was analysed using an ATR infrared spectrophotometer (no difference was found whether the sample was left to dry on the spectrophotometer, usually within a few minutes, or if it was dried in a desiccator prior to infrared analysis). This control experiment was used to confirm β-haematin formation under the reaction conditions. The effect of each compound was determined by adding three equivalents to the test tube prior to the addition of the acetate buffer solution. In each run a negative inhibition control (haemin alone) and a positive inhibition control (a reaction mixture containing three equivalents of chloroquine phosphate) were used. The formation of β-haematin may be determined by the appearance of two peaks on the infrared spectrum of solid residue, one at 1660 cm-1 and one at 1207 cm-1.30
Acknowledgements
The authors would like to thank Dr. Kirsten Barnes (UKZN) for the data collection of the copper and iron terpyridyl complexes. We would also like to express our gratitude to Prof. Timothy Egan (UCT) for his helpful suggestions in preparation of this manuscript. The NRF is gratefully acknowledged for its funding of this work.
References and Notes
1 WHO World Malaria Report 2005, Roll Back Malaria, UNICEF pp xi-xvii, UNICEF. [ Links ]
2 J.A. Ocheskey, V.R. Polyakov, S.E. Harpstrite, A. Oksman, D.E. Goldberg, D. Piwnica-Worms and V Shama, J. Inorg. Biochem., 2003, 93, 265-270. [ Links ]
3 D. Rasoloson, L.R. Shi, C.R. Chong, B.F. Kafsack, D.J. Sullivan, Biochem J., 2004, 381, 803-811. [ Links ]
4 R. Sanchezlopez and K. Haldar, Mol. Biochem. Parasitol, 1992, 55, 9-20. [ Links ]
5 S.J. Oppenheimer, F.D. Gibson, S.B. Macfarlane, J.B. Moody, C. Harrison, A. Spencer and O. Bunari, Trans. Royal Soc. Trop. Med. Hyg., 1986, 80, 603-612. [ Links ]
6 T.E.A. Peto and J.L. Thompson, Br. J. Haematology, 1986, 63, 273-280. [ Links ]
7 S. Pollack and V Schnelle, Br. J. Haematology, 1988, 68, 125-129. [ Links ]
8 P.W.J. Harvey, P.F. Heywood, M.C. Nesheim, K. Galme, M. Zegans, J.P Habicht, L.S. Stephenson, K.L. Radimer, B. Brabin, K. Forsyth and M.P. Alpers, Am. J. Trop. Med. Hyg., 1989, 40, 12-18. [ Links ]
9 L.W. Scheibel and G.G. Stanton, Mol. Pharmacol., 1986, 30, 364-369. [ Links ]
10 M. Loyevsky, S.D. Lytton, B. Mester, J. Libman, A. Shanzer and Z.I. Cabantchik, J. Clin. Invest., 1993, 91, 218-224. [ Links ]
11 P.J. Rosenthal and S.R. Meshnick, Mol. Biochem. Parasitol., 1996, 83, 131-139. [ Links ]
12 T.J. Egan, J.M. Combrinck, J. Egan, G.R. Hearne, H.M. Marques, S. Ntenteni, B.T. Sewell, P.J. Smith, D. Taylor, D.A. van Schalkwyk and J.C. Walden, Biochem. J., 2002, 365, 343-347. [ Links ]
13 G. Lowe, Great Britain, 2000, Vol. 2000-GB686. [ Links ]
14 A. Albert, S.D. Rubbo and M.I. Gibson, Br. J. Exp. Pathol., 1950, 31, 425-441. [ Links ]
15 A. Albert, S.D. Rubbo and M.I. Gibson, Br. J. Exp. Pathol., 1953, 34, 119-130. [ Links ]
16 D.S. Sigman, A. Mazumder and D.M. Perrin, Chem. Rev., 1993, 93, 2295-2316. [ Links ]
17 K.W. Jennette, S.J. Lippard, Vassilia Ga and W.R. Bauer, Proc. Natl. Acad. Sci. U.S.A., 1974, 71, 3839-3843. [ Links ]
18 L. Messori, F. Abbate, G. Marcon, P. Orioli, M. Fontani, E. Mini, T. Mazzei, S. Carotti, T. O'Connell and P. Zanello, J. Med. Chem., 2000, 43, 3541-3548. [ Links ]
19 G. Lowe, A.S. Droz, T. Vilaivan, G.W. Weaver, L. Tweedale, J.M. Pratt, P. Rock, V. Yardley and S.L. Croft, J. Med. Chem., 1999, 42, 999-1006. [ Links ]
20 A. Kitjaroentham, T. Suthiphongchai and P. Wilairat, Acta Trop., 2006, 97, 5-9. [ Links ]
21 G.E.M. Maguire, The datafor the copper terpyridyl complex has been deposited in the Cambridge database CCDC 762560. [ Links ]
22 G.E.M. Maguire, The data for the Iron complex has been deposited in the Cambridge database CCDC 762561. [ Links ]
23 H.B. Friedrich, G.E.M. Maguire, B.S. Martincigh, M.G. McKay and L.K. Pietersen, Acta Crystallogr., Sect. E-Struct. Rep. Online., 2009, 65, E13-E13. [ Links ]
24 S. Bonse, J.M. Richards, S.A. Ross, G. Lowe and R.L. Krauth-Siegel, J. Med. Chem., 2000, 43, 4812-4821. [ Links ]
25 S.A. Ross, C.A. Carr, J.W. Briet and G. Lowe, Anti-Cancer Drug Design., 2000, 15, 431-439. [ Links ]
26 K. Becker, C. Herold-Mende, J.J. Park, G. Lowe and R.H. Schirmer, J. Med. Chem., 2001, 44, 2784-2792. [ Links ]
27 R. Ahmadi, S. Urig M. Hartmann, B.M. Helmke, S. Koncarevic, B. Allenberger, C. Kienhoefer, M. Neher, H.H. Steiner, A. Unterberg C. Herold-Mende and K. Becker, Free Radical Biol. Med., 2006, 40, 763-778. [ Links ]
28 T.J. Egan and H.M. Marques, Coord. Chem. Rev., 1999, 192, 493-517. [ Links ]
29 A.F.G. Slater and A. Cerami, Nature, 1992, 355, 167-169. [ Links ]
30 T.J. Egan, D.C. Ross and P.A. Adams, FEBS Lett., 1994, 352, 54-57. [ Links ]
31 T.J. Egan, K.R. Koch, P.L. Swan, C. Clarkson, D.A. Van Schalkwyk and P.J. Smith, J. Med. Chem., 2004, 47, 2926-2934. [ Links ]
32 B.S. Furniss, A.J. Hannaford, P.W.G. Smith and A.R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, Longman Scientific & Technical copublished with John Wiley and Sons, New York, 1989. [ Links ]
33 L.S. Hollis and F.H. Burnstall, J. Chem. Soc., 1934, 1498-1500. [ Links ]
34 L.S. Hollis and S.J. Lippard, J. Am. Chem. Soc., 1983, 105, 4293-4299. [ Links ]
35 A.C. Sant'Ana, W.A. Alves, R.H.A. Santos, A.M.D. Ferreira and M.L.A. Temperini, Polyhedron, 2003, 22, 1673-1682. [ Links ]
36 D.J. Hathcock, K. Stone, J. Madden and S.J. Slattery, J. Inorganica Chimica Acta, 1998, 282, 131-135. [ Links ]
37 R. Karkalic and Z.D. Bugarcic, Monatsh. Chem, 2000, 131, 819-824. [ Links ]
Received 21 December2012
Revised 7 March 2013
Accepted 12 March 2013
* To whom correspondence should be addressed. E-mail: maguireg@ukzn.ac.za

