










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban ago. 2013
RESEARCH ARTICLE
Negishi coupling of pteridine-O-sulfonates
Winston NxumaloI, *; Andrew DinsmoreII
IDepartment of Chemistry, University of Limpopo Turfloop Campus, Limpopo, Private Bag X1106, Sovenga, 0727, South Africa
IISchool of Chemistry, University of the Witwatersrand, Johannesburg, Private Bag X3, P.O. WITS, 2050, South Africa
ABSTRACT
Negishi coupling of pteridine-O-sulfonate with Zn-aryls is reported. Hydrolysis of the protecting groups with 1M NH3 gave the 6-subustituted 2,4-diaminopteridine while hydrolysis with 1 M NaOH gave the 6-subsituted pterin.
Keywords: Pteridine, pterin, Negishi coupling.
1. Introduction
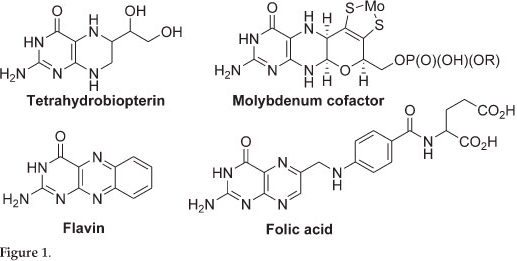
Pterins are incorporated into a number of redox active cofactors which are essential to all forms of life, examples include tetrahydrobiopterin (BH4), the molybdenum cofactors (Moco), flavin and folic acid (Fig. 1). Deficiency in these essential cofactors results in severe birth defects and in most cases death. The first total synthesis of pterins was reported in the 1940s and since then a number of naturally occurring pterins and their derivatives have been synthesized.1,2 However, most of the synthesized pterins are folic acid derivatives and few synthetic routes in making non-folic acid derivatives exist.
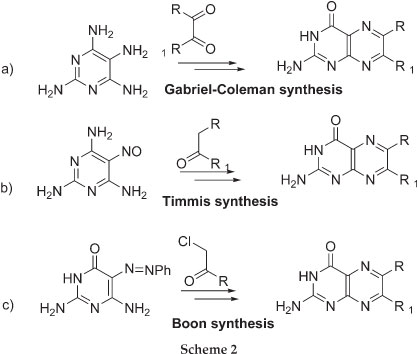
The current methods of preparing non-folic acid derivatives include the preparation of 6-chloropterin which can undergo nucleophilic substitution and Sonogashira coupling reactions (Scheme 1).3 Other methods include the condensation of pyrimidone or pyrimidine ring with a substituted glyoxal, α-carbonylmethylene group and α-haloketones to give the corresponding 6-substituted pterin and pteridine rings, respectively (Scheme 2).4-6 A number of natural pterin compounds have been synthesised following these procedures but, a number of problems such as selectivity of 6- vs. 7-position and difficulty with certain chemical transformations, have been reported.

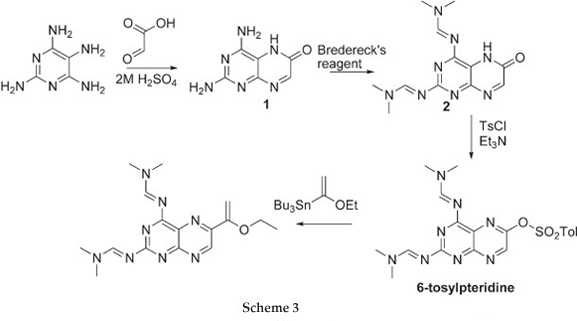
The challenge now lies in developing a synthetic method which is selective and versatile. Work reported by Joule et aU demonstrated the preparation of 6-tosyloxypteridine, selectively, in three steps starting from tetra-aminopyrimidine and glyoxylic acid. The 6-tosyloxypteridine was subsequently shown to undergo Stille coupling reaction (Scheme 3). We have recently demonstrated Sonogashira coupling reactions on 6-benzene-sulfonatepteridine, selective hydrolysis to give either the 6-subustituted 2,4-diaminopteridine or 6-substituted pterin.8
In this paper we report Negishi coupling reactions on 6-benzenesulfonatepteridine, selective hydrolysis of the protecting groups to give exclusively the 6-substituted 2,4-diamino-pteridine and 6-substituted pterins.
2. Results and Discussion
Treating 2,4-diaminopyrimidine and glyoxylic acid in 2 M H2SO4 gave 2,4-diaminopterin-6-one 1 in 74 % yield as described by Pfleiderer et al9 The addition of Bredereck's reagent10 afforded the more soluble 2,4-Di(N,N-dimethylamino-methyleneamino)pteridine-6-one7 2 in 91 % yield which was treated with benzenesulfonylchloride to give 2,4-Di(N,N-dimethylaminomethyleneamino)-6-benzenesulfonyloxy-pteridine 3 in 75 % yield.8
Previous work reported on the 6-benzenesulfonatepteridine showed the amidine protecting group to be easily removed by mild acidic or basic conditions,7-8 and this was a potential challenge given that a stoichiometric amount of an organo-metallic reagent would be required for the Negishi coupling reaction. Another challenge faced was the fact that the pteridine-6-sulfonate 3 was not soluble in most solvents used for Negishi coupling reactions.
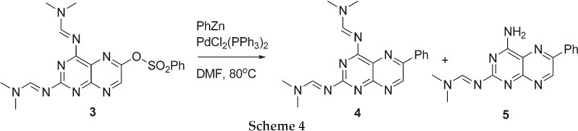
We began our investigation by treating 3 with phenyl-Zn under Negishi coupling conditions in refluxing THF, but after an hour a bulky precipitate was observed. HPLC-MS analysis of both the supernatant and the precipitate showed presence of starting material along with deprotected starting material. When changing the solvent to DCM, no precipitates were observed but only unreacted starting material was obtained. These results demonstrated that the organometallic reagent was not interfering with the amidine protecting group. We then turned our attention to a polar aprotic solvent, DMF, cautious of the fact that it can act as an electrophile towards the phenyl-Zn reagent. The 6-benzenesulfonatepteridine 3 was treated with phenyl-Zn under Negishi coupling conditions in DMF at 80 °C for two hours and, to our delight, the coupled phenyl-pteridine 4 along with the mono-deprotected phenyl-pteridine 5 was formed. Work by Joule et al.7 also reported the product mixture of desired Stille coupling product and mono-deprotected product, and recently we reported a mixture of Sonogashira coupling products.8 Further purification of the two close running products, by flash chromatography, gave a product distribution that favoured the mono-deprotected product 5 in ~1:9 ratio in 80 % overall yield. To our knowledge, no Negishi coupling reactions have been reported on pteridine ring systems.
Having shown that 6-benzenesulfonatepteridine 3 can undergo Negishi coupling reactions, we investigated varying the aryl groups. Treating 3 with zincyl-trimethoxybenzene under our optimized Negishi coupling conditions gave a mixture of two products which we assigned to be the desired coupling product 6 along with the mono-deprotected product 7 in ratio (Fig. 2). Further purification gave a product distribution in favour of 7 in ~1:9 ratio in 65 % overall yield. Similarly, treating 3 with 2-zincyl-furan gave a mixture of two products which we assigned to be the desired coupling product 8 along with the mono-deprotected product 9 in ratio (Fig. 2) and further purification gave a product distribution favouring 9.

We then investigated the ability of mesylate as a leaving group in pteridine system, and treated the mesyloxy-pteridine 10 with phenyl-Zn under the coupling conditions and obtained the mixed coupled phenyl-pteridines 4 and 5 in 78 % yield. However, the attempted coupling of the zincated-trimethoxyphenyl and furan rings with the mesyloxy-pteridine 10 was unsuccessful. This could be due to the presence of lithium (Li) ions in solutions that have been reported to be poisonous to the palladium catalyst in certain cross-coupling reactions.10 The furan-2-ylzinc and trimethoxyphenylzinc are prepared by transmetallation from their corresponding lithio-derivatives and are never purified meaning that Li ions are still present, in stoichiometric amounts, in solution during the Negishi coupling reaction. The extent of poisoning by Li and sometimes Mg is not fully understood and is also dependant of the substrates attached to the zinc metal and also the leaving group.11
The attention then shifted to the hydrolysis of the amidine protecting group. In recent studies we demonstrated that under mild hydrolytic conditions the 2,4-diaminopteridine is obtained, and under stronger hydrolytic conditions the corresponding pterin is obtained.8 Treating the phenyl-pteridine 4 and 5 with 1MNH3 solution under gentle reflux gave 2,4-diamino-6-pteridine1211 in 60 % yield. Hydrolysis with 1 M NaOH/EtOH solution gave 6-phenyl-pterin1212 in 61 % yield (Scheme 5).

When treating the trimethoxyphenyl-pteridines 6 and 7 with 1MNH3 solution under gentle reflux, we obtained 6-tri-methoxy-pterin 13 instead of the desired 2,4-diaminopteridine analogue. Similar results were obtained with the furyl-pteridines 8 and 9 where 6-furyl-pterin 14 was obtained (Fig. 3).

These results suggest that the groups attached at the 6-position have a direct effect in the reactivity of the pterin ring. When electron rich substituents, relative to phenyl, are attached at the 6-position, the hydrolysis of the 4-amino group is easily achieved under weak basic conditions such as ammonia. It is likely that the substituents changes the electron density of the ring and therefore favour the addition of weak nucleophile (water) and the loss of ammonia gives the pterin ring.
In conclusion, we have demonstrated that 6-benzene-sulfonatepteridine 3 is a useful substrate for Negishi coupling reactions.
3. Experimental
Synthesis of 2,4-Diaminopteridin-6-one (1)9
Tetraaminopyrimidine (21.17 g, 0.136 mol) was added to 2 M H2SO4 (550 mL), pre-heated to 80 °C and solution stirred for 10 min. Glyoxylic acid (16.35 g, 0.272 mol, 2 eq.) was added, the solution stirred for a further 15 min then allowed to cool to room temperature and left to stir for 2 h. The resultant suspension was filtered, and the mustard solid washed with water (3 x 50 mL). The solid was suspended in water (150 mL) and neutralized with sat. NaHCO3 (cautiously added in 10 mL portions until effervescence stopped, ca. ~150 mL was required), the solid filtered and washed with water (5 x 60 mL) then MeOH (3 x 50 mL), air-dried, and then dried under reduced pressure over P2O5 to give 2,4-diaminopteridin-6-one (17.952 g, 74 %) as a mustard solid. m.p. (dec.) >300 °C; m/z (ESI) 179 (M+H+, 100); λmax (1 M NaOH) 410 and 265 nm. Physical and spectroscopic data agree with those reported elsewhere.9
2,4-Di(N,N-dimethylaminomethyleneamino)pteridine-6-one (2)
2,4-Diaminopteridin-6-one (4.02 g, 0.0225 mol) and Bredereck's reagent10 (14 mL, 0.0675 mol, 3 eq.) were stirred together in DMF (50 mL), under an Ar atmosphere and warmed to 65 °C for 3 h, and allowed to cool to room temperature. The yellow precipitates were filtered, washed with little cold DMF then Et2O, air-dried and finally dried under reduced pressure over P2O5 to give 2/4-di(N/N- imethylaminomethyleneamino) pteridine-6-one (6.20 g, 91 %); m.p. (dec.) 270 °C; δH (300 MHz, CDCl3) 3.07 (3H, s), 3.08 (3H, s), 3.10 (3H, s), 3.15 (3H, s), 8.15 (1H, s), 8.48 (1H, s) and 8.76 ppm (1H, s); m/z(EI) 288 (M+, 100), 273 (45), 244 (19) and 232 (45). Spectroscopic data agree with those reported elsewhere.7
3.3. 2,4-Di(N/N-dimethylaminomethyleneamino)-6-benzene-sulfonyloxypteridine (3)
2,4-Di(N/N-dimethylaminomethyleneamino)pteridine-6-one (4.160 g, 14.5 mmol), DMAP (0.2 g, 1.45 mmol) and benzene sulfonyl chloride (3.8 mL, 29 mmol, 2 eq.) were dissolved in DCM (40 mL) and cooled to 0 °C. Et3N (3.7 mL, 29 mmol, 2 eq.) was added drop-wise into the mixture, and the final solution was allowed to warm to room temperature and stirred for 1 h. The reaction was quenched with sat. NaHCO3 (15 mL), the organic layer separated and the aqueous layer washed with DCM (2 x 20 mL). The combined organic layers were dried over MgSO4, and the solvent evaporated to dryness to give a dark yellow solid which was recrystalized from EtOH to give 2,4-di(N,N-dimethylaminomethyleneamino)-6-benzenesulfonyloxopteridine (4.62 g, 75 %) as a yellow solid; m.p. (dec) 203 205 °C; δH (CDCl3, 300 MHz) 3.19 (3H, s), 3.22 (3H, s), 3.23 (3H, s), 3.30 (3H, s), 7.53 7.79 (3H, m), 8.25 8.28 (2H, dd, J = 7.8,0.8 Hz), 8.56 (1H, s) and 8.95 8.97 ppm (2H, overlapping singlet); dC (CDCl3, 100 MHz) 35.4, 41.4, 125.2, 129.0, 129.4, 134.4, 136.3, 144.4, 148.6, 155.6, 158.3,159.8,166.5 and 168.5 ppm; m/z (EI) 428 (M+, 21 %), 413 (40), 288.2 (100), 273.1(45), 232 (40), 217 (80) and 162.9 (35) (HREI found: 428.13775. C18H20N8O332S requires 428.13791).
General Method for Negishi Coupling
To the solution of Zn-reagent (3.15 mmol) in DMF/THF mixture (15 mL, 2:1) was added benzenesulfonyl-pteridine 3 (2.1 mmol, 900 mg), Pd2(dba)3CHCl3 (125 mg, 0.1575 mmol, 5 mmol %) and PPh3 (125 mg, 0.477 mmol). The final solution was warmed to 80 °C and stirred under Ar for 2 h, concentrated on a rotary evaporator and purified on flash silica, eluting with 5 % MeOH/DCM.
4-Amino-2-(N,A/-dimethylaminomethyleneamino)-6-(phenyl)pterin (5)
Phenyl-ZnCl (3.15 mmol) was treated with 3 according to general method to give 850 mg of mixed 4 and 5. Further purification on flash silica, eluting with 5 % MeOH/DCM gave 4-amino-2-(N,N-dimethylaminomethyleneamino)-6-(phenyl)pterin as an orange solid (494 mg, 80 %); Rf0.28; m.p. 180 °C; dH (d6-DMSO, 300 MHz) 3.08 (3H, s), 3.19 (3H, s), 7.51 7.53 (3H, m), 8.418.43 (2H, m), 8.76 (1H, s), 9.60 (1H, s); m/z (APCI) 294 (M + H+, 100), 239 (80)(HRESI +ve mode Found: 294.14736, C15H16N7 requires 294.14620).
4-Amino-2-(N,A/-dimethylaminomethyleneamino)-6-(3,4,5-trimethoxyphenyl)pterin (7)
3,4,5-Trimethoxyphenyl-ZnCl (3.15 mmol) was treated with 3 according to the general method to give 672 mg of mixed products 6 and 7. Further purification on flash silica, eluting with 5 % MeOH/DCM gave 4-amino-2-(N,N-dimethylaminomethylene-amino)-6-(3,4,5-trimethoxyphenyl)pterin as an orange solid (520 mg, 65 %); Rf 0.25; m.p. 160 °C; δH (d6-DMSO, 300 MHz) 3.08 (3H, s), 3.18 (3H, s), 3.74 (3H, s), 3.94 (6H, s), 7.66 (2H, s), 8.76 (1H, s), 9.59 (1H, s); m/z (APCI) 384 (M + H+, 100), 329 (40) (HRESI +ve mode found: 384.17772, C18H22N7O3 requires 384.17786).
4-Amino-2-(N,N/-dimethylaminomethyleneamino)-6-(furan-2-yl)pterin (9)
Furan-2-yl-ZnCl (3.15 mmol) was treated with 3 according to the general method to give 700 mg of mixed products 8 and 9. Further purification on flash silica, eluting with 5 % MeOH/ DCM gave 4-amino-2-( N,N-dimethylaminomethyleneamino)-6-(furan-2-yl)pterin as a dark-red solid (400 mg, 67 %); Rf 0.27; m.p.(dec) 300 °C; δH (d6-DMSO, 300 MHz) 3.06 (3H, s), 3.16 (3H, s), 6.75 (1H, m), 7.31 (1H, m), 7.42 (1H, m), 8.73 (1H, s) and 9.22 (1H, s); m/z (APCI) 284 (M + H+, 100), 229 (30) (HRESI +ve mode Found: 284.1232, C13H14N7O) requires 284.1254).
2,4-Diamino-6-(phenyl)pteridine (11)12
4-Amino-2-(N,N-dimethylaminomethyleneamino)-6-(phenyl) pterin 5 (150 mg, 0.512 mmol) was treated with 1 M NH3/EtOH (8 mL, 1:1) and gently refluxed for 8 h, allowed to cool to room temperature, acidified with AcOH and placed in a fridge (2 °C) for 18 h. The solution was filtered, crystals washed sequentially with water, EtOH and Et2O to give 2,4-diamino-6-(phenyl)pteridine as a dark green solid (70 mg, 57 %); m.p. >300 °C; δH (d6-DMSO, 300 MHz) 7.50 (3 H, m), 8.18 (2 H, m) and 9.30 (1 H, s); m/z (APCI) 239 (M + H+, 100) (HRESI Found: 239.1046, C12H11N6 requires 239.10397). Physical and spectroscopic data agree with those reported elsewhere.11
6-(Phenyl)pterin (12)12
4-Amino-2-(N,N-dimethylaminomethyleneamino)-6-(phenyl) pterin 5 (150 mg, 0.512 mmol) was treated with 1M NaOH/EtOH (8 mL, 1:1) and gently refluxed for 8 h, allowed to cool to room temperature, acidified with AcOH and placed in a fridge (2 °C) for 18 h. The solution was filtered, crystals washed sequentially with water, EtOH and Et2O to give 6-(phenyl)pterin as a yellow solid (60 mg, 61 %); m.p. >300 °C; λmax (nm) 296, 369; δH (d6-DMSO, 300 MHz) 7.51 (3 H, m), 8.12 (2 H, m) and 9.26 (1 H, s); m/z (APCI) 239 (M+, 100). Physical and spectroscopic data agree with those reported elsewhere.11
6-(3,4,5-Trimethoxyphenyl)pterin (13)
4-amino-2-(N/N-dimethylaminomethyleneamino)-6-(3,4,5-tri methoxyphenyl)pterin7 (150 mg, 0.391 mmol) was treated with 1M NaOH/EtOH (8 mL, 1:1) and gently refluxed for 8 h, allowed to cool to room temperature, acidified with AcOH and placed in a fridge (2 °C) for 18 h. The solution was filtered, crystals washed sequentially with water, EtOH and Et2O to give 6-(3/4/5-trimethoxyphenyl)pterin as a green-yellow solid (74 mg, 59 %); m.p. >300 °C; δH (d6-DMSO, 300 MHz) 3.72 (3 H, s), 3.89 (6 H, s), 7.38 (2 H, s) and 9.23 (1 H, s); λmax (nm) 305, 379; m/z (APCI) 330 (M+H+). Attempts to get high resolution mass measurements were unsuccessful in both HREI and HRESI instruments.
6-(Furan-2-yl)pterin (14)
4-amino-2-(N/N-dimethylaminomethyleneamino)-6-(furan-2-yl)pterin 9 (150 mg, 0.530 mmol) was treated with 1 M NaOH/EtOH (8 mL, 1:1) and gently refluxed for 8 h, allowed to cool to room temperature, acidified with AcOH and placed in a fridge (2 °C) for 18 h. The solution was filtered, crystals washed sequentially with water, EtOH and Et2O to give 6-(furan-2-yl)pterin as a brown solid (76 mg, 63 %); m.p. >300 °C; δH (d6-DMSO, 300 MHz) 6.68 (1 H, apparent s), 7.14 (1 H, apparent s), 7.86 (1 H, apparent s) and 8.97 (1H, s); λmax (nm) 309, 395; m/z (APCI) 230 (M + H+, 100). Attempts to get high resolution mass measurements were unsuccessful in both HREI and HRESI instruments.
References
1 D.J. Brown, Heterocyclic Compounds/ Fused Pyrimidines, John Wiley and Sons, 1988, 1-4. [ Links ]
2 R. Purrmann, Justus Leibigs Ann. Chem./1940, 544, 182. [ Links ]
3 E.C. Taylor and R. Kobylecki, J. Org. Chem./ 1978, 43, 680. [ Links ]
4 T. Hanaya, H. Baba and H. Yamamoto, Carbohydrate Res./ 2007, 342, 2159. [ Links ]
5 S. Goswami and A.K. Adak, Tetrahedron Lett./ 2002, 43, 8371. [ Links ]
6 E.C. Taylor, K.L. Perlman, I.P. Sword, M. Sequin-Frey and P.A. Jacobi, J. Am. Chem. Soc./ 1973, 95, 6407. [ Links ]
7 A. Dinsmore, D.C. Garner, and J.A. Joule, Tetrahedron/ 998, 54, 9559. [ Links ]
8 W. Nxumalo and A. Dinsmore, Heterocycles/ DOI: 10.3987/COM-12-12610. [ Links ]
9 G. Konrad, and W. Pfleiderer, Chem. Berr., 1970, 103, 735. [ Links ]
10 H. Bredereck, G. Simchen, H. Hoffman, P. Horn and R. Wahl, Angew. Chem./ 1967, 79, 311. [ Links ]
11 E. Negishi, Q. Hu, Z. HuangM., Qian and G. Wang, Aldrichimica Acta/ 2005, 38(3), 71. [ Links ]
12 E.C. Taylor, K.L. Perlman, Y-H. Kim, I.P. Sword and P.A. Jacobi, J. Am. Chem. Soc./ 1973, 95(19), 6413. [ Links ]
Received 1 November 2012
Revised 2 December 2012
Accepted 6 December 2012
* To whom correspondence should be addressed. E-mail: winston.nxumalo@ul.ac.za

