










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
RESEARCH ARTICLE
Quantification of rhodium in a series of inorganic and organometallic compounds using cobalt as internal standard
Trevor T. Chiweshe; Walter Purcell*; Johan A. Venter
Department of Chemistry, University of the Free State, 205 Nelson Mandela Avenue, Bloemfontein, 9300, South Africa
ABSTRACT
An analytical method for the quantification of rhodium using inductively coupled plasma optical emission spectrometry (ICP-OES) and cobalt as internal standard was developed. Rhodium recovery was determined in different samples, which included a certified reference material (CRM), pure rhodium metal, inorganic RhCl.3H2O salt as well as different organometallic compounds. Excellent rhodium recoveries of 100,100, 99.0 and 99.7 % with relative standard deviations (RSD) of 1.0,1.0, 0.3 and 0.4 % were obtained for the CRM, rhodium metal sample, RhCl3·3H2O and [Rh(cupf)(PPh3)(CO)(CH3)(I)], respectively. The accurate recovery of rhodium was, however, affected by the presence of easily ionized elements (EIE) and unmatched acid matrixes with a decrease in rhodium recovery of up to 16 %. Validation parameters such as accuracy, precision, specificity, limit of detection (LOD) and robustness were all investigated to confirm the suitability of this newly developed analytical procedure for rhodium determination.
Key words: Rhodium, ICP-OES, cobalt internal standard, quantification.
1. Introduction
Rhodium is a member of the platinum group metals (PGM), which include the lighter elements palladium and ruthenium and the heavier triad platinum, iridium and osmium. The PGM in South Africa also includes gold and these seven elements are currently considered to be the most expensive or precious metals on the open market with values ranging from $1642 oz-1 for gold, $1599 oz-1 for platinum to $116 oz-1 for ruthenium.1 Factors such as their low natural abundance in the earth's crust (0.001 g ton-1), PGM concentrations ranging between 3.0 and 5.5 g ton-1 in the well-known Plat- and Merensky Reef deposits2 and localized worldwide distribution contribute to the high economic value of these metals.
Rhodium has a number of interesting applications in industry, ranging from the manufacturing of expensive jewellery to the use as an alloying agent for hardening and improving the corrosion resistance3 of platinum and palladium. These alloys are used in furnace windings, bushings for glass fibre production, electrodes for aircraft spark plugs, laboratory crucibles4 and thermocouple elements. A wire alloyed with 10 % rhodium and 90 % platinum forms an excellent thermocouple for measuring high temperatures in an oxidizing atmosphere.5 The use of rhodium, platinum and palladium in the so-called three-way catalyst in the control of toxic emissions from automotives contribute significantly to the worldwide consumption of the three metals.
The element also exhibits scope and versatility as a homogeneous catalyst in the production of a number of organic compounds on industrial scale, which is apparently unmatched by any other PGM6 or metal catalyst. Among the rhodium-catalyzed reactions that have received significant attention are the hydrogenation of olefins,7 including the first commercial asymmetric catalytic process (synthesis of L-3,4-dihydroxyphenylalanine, for the treatment of Parkinson's disease),8,9 hydrogenation of arenes,10 hydroformylation of olefins and olefin diene codimerization.11 Rhodium used in catalysts such as rhodium hydridocarbonyl tristriphenylphosphine, [HRh(CO)(PPh3)3], for the hydroformylation of olefins must be reactivated periodically and/or recovered from the by-products for re-use (U.S. Patent 4,390,473).
In 1992, Brooks reported that 'Platinum group metals (PGM), e.g. Rh, Pd, Os, Pt, Ir and Ru, present the ultimate challenge to the analytical chemistry researcher'.12 The high intrinsic value of this group of elements necessitates analytical methods and tools that are able to analyze with high precision (RSD levels of 3-5 %) and accuracy at extremely low levels of PGM concentration (low ppm range) in samples, which include PGM minerals, metal alloys, road dust, soils, airborne particles, waste water and biological matter. Several challenges exist in the development of analytical techniques that adhere to accuracy and precision at trace level PGM quantification. These challenges include low and non-homogenized PGM concentration levels, spectral interferences due the presence of oxides and double-charged ions, as well as the complex chemical behaviour and physical similarities of the PGM. Accuracy of rhodium determination is also hampered by insufficient sensitivity of existing instruments, interference derived from easily ionizable elements (EIE), acid matrix, chemical and spectral interferences13-15 and low detection limits in spectrometric techniques. Inferior sensitivity and poor selectivity16 at trace levels cause unsatisfactory recoveries (67-87 %) using these techniques.17
Greenfield et al.18 reported that analyte emission signal reduction is caused by high acid concentrations leading to a reduction of analyte transport into the plasma. The degree of depression is also dependent on the acid type.19-24 Results obtained from these studies also indicted that variations in the solution density and viscosity alter the venturi aspiration rate, which resulted in a change in the sensitivity of the equipment. Other factors such as aerosol generation efficiency, droplet size distribution and aerosol transport losses play a role in the variation of analytical results.25,26 Acid matrix and interferences derived from the EIE have a profound effect on the accuracy of PGM trace elemental analysis. Inorganic mineral acids such as HNO3, HF, HCl and HBr27,28 and alkali solutions such as the soluble alkali metal hydroxides, LiOH, NaOH, KOH and CsOH are often used for sample dissolution and for maintaining the stability of the analyte(s) solution.29,30 Excess acid or alkali solutions and the presence of EIE can be a source of error in the rhodium determination. Several methods have been proposed to compensate for these interferences and the techniques include the stabilization of solution uptake rate by a peristaltic pump, introduction of an internal reference element, formal mathematical methods, matrix matching techniques, standard addition method and the use of correction factors.9,31,32 Most matrix correction approaches involve matrix matching, internal standard and standard addition methods. However, matrix matching is seldom applied in situations where the sample matrix is poorly defined.
Spectrometric techniques such as inductively coupled plasma optical emission spectroscopy (ICP-OES33, mass spectroscopy (ICP-MS), atomic absorption spectroscopy (AAS)34 and X-ray fluorescence (XRF)35 have lately been employed for the accurate determination of rhodium. Several ISO-approved ICP-OES methods, using yttrium as internal standard, are described for the precious metal determination in metal alloys, while gold and gold alloy analysis requires yttrium or indium as internal standards. The ICP-OES has become the dominant method for rapid spectroscopic multi-element analyses due to low detection limits, wide linear dynamic range, and high precision and selectivity. Internal standardization of ICP-OES measurements is also successful in increasing the accuracy and precision of PGM analysis due to the reduction of plasma noise36,37 instrumental drifts and matrix interferences.
Questions still remain about the accurate determination of rhodium in the electroplating and precious metal refining industries.38 Low rhodium recoveries in organometallic complexes (81-96 %) also illustrates the uncertainty of rhodium quantification39-41 and is probably due to unreliable methods and techniques. The main objective of this research was to evaluate cobalt as internal standard for the accurate determination of rhodium in different samples, which included a rhodium CRM, pure metal (RM), inorganic salts and different organometallic compounds. The investigation included a comparison with external, standard addition and yttrium internal standard calibration results. The robustness of this method was evaluated by its ability to compensate for instrumental drifts, background emission, acid matrix and matrix effects due to the presence of EIEs.
2. Instrumentation
2.1. ICP-OES
A Shimadzu ICPS-7510 ICP-OES with a radial-sequential plasma spectrometer was used for the wet chemical analysis of all the samples that were investigated. The vertically oriented ICP-OES with the 'radial viewing' plasma was chosen to be suitable for the analysis due to its better detection limits, compared to the axial viewing plasma. Default conditions were used as indicated in Table 1 in order to achieve the best precision and accuracy of results.

2.2. Microwave Digestion
An Anton Paar Perkin-Elmer Multiwave 3000 microwave digestion system equipped with an 8SXF 100 rotor and eight polytetrafluoroethylene (PTFE) reaction vessels were used for the acid dissolution of the powdered rhodium metal and the CRM (European Reference Material ERM®-504) samples. An internal program for the digestion of the platinum group metals (PGM XF100-8) was selected with conditions as set in Table 2.
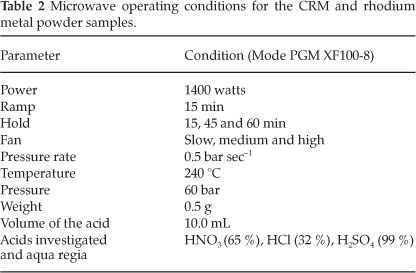
3. General Experimental Procedures
Double-distilled water, using an electronic distillatory vessel (Fisons w/FF9/4), was used for a ll the analytical solution preparations. All the samples were weighed accurately to 0.1 mg at 25 ° C using a Scaltec (SBA 33) electronic balance, which was tested under ISO 9001. A Transferpette micro-pipette (±0.5 µL) was used for the accurate measurement and transfer of the different liquids and acids at 25 °C. The presence of any interfering elements in the rhodium CRM and samples was verified with a qualitative scanning program incorporated in the ICP-OES software. Their presence was monitored at the most intense emission wavelength of each element. Elements were considered present if they exhibited strong emission at three of the chosen wavelengths. Elements that did not exhibit all the characteristic lines were considered to be entirely absent or below the instrument's limit of detection. All ICP-OES results given are the average of the three individual replicate readings taken sequentially. All the calculations for the rhodium percentage recoveries were made at a 95 % confidence interval, which was adopted from the CRM (European Reference Material ERM®-504).
4. Reagents and Glassware
The different standard solutions (1000.0 ppm rhodium and yttrium in 2-3 % HNO3), cobalt nitrate (Co(NO3)2-6H2O) (99.99 % assay) and the rhodium metal powder (99.99 %) were purchased from Sigma-Aldrich while all the other chemicals used in this study from Merck Chemicals. All the chemicals and reagents such as ethanol, acetone, acetylacetone (acac), cupferron (cupf), diphenyl-2-pyridylphosphine (DPP) and N,N-dimethyl-formamide (DMF) were used without further purification, except for triphenylphoshine (PPh3),which was further purified by recrystallization in methanol prior to use. The beakers and volumetric flasks used in this research were of the Schott Duran type and the volumetric flasks were of Blaubrand, grade (A) type.
5. Description of the CRM
The CRM used in this study was the European Reference Material (ERM®-504) which was purchased from Germany and certified by the Bundesanstalt für Materialforschung und Prüfung (BAM) in cooperation with the Committee of Chemists of the GDMB, Gesellschaft für Bergbau, Metallurgie, Rohstoff und Umwelttechnik. The ERM®-504 sample was obtained as a mixture ofused automobile catalysts and was supplied and prepared by a commercial manufacturer. According to the certificate, the material was ignited, ground to a particle size of less than 100 µm and was homogenized thoroughly before bottling. The certified and the uncertainty values (in brackets) of platinum, palladium and rhodium in the ERM®-504 were reported as 1777(±15), 279(±6) and 338(±4) mg kg-1 respectively.
6. Experimental
6.1. Wavelength Selection and the Determination of Detection and Quantification Limits of Rhodium
The selection of the most suitable wavelengths for rhodium, yttrium and cobalt analysis was done using the ICP-OES 'profile' function as discussed in Section 7.1. The program allows for a rapid semi-quantitative analysis for a multiple wavelength analysis and records the number of scans of an analyte solution and that of the matrix at low concentration. The spectra are then superimposed on each other and the resultant spectrum is a combination of all possible spectral lines of the elements present in the sample (CRM) and of rhodium in this case. This allows for the selection of the wavelength of the analyte with little or no interference from the other elements, as shown in Fig. 1.
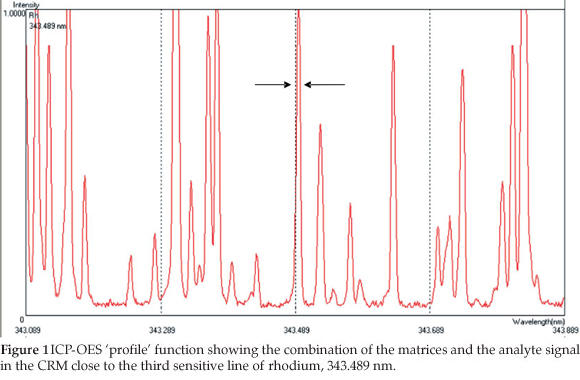
The rhodium atomic line at 343.489 nm, the yttrium ionic line at 377.433 nm and the cobalt ionic line 228.616 nm were chosen for the analysis. Calibration standards were prepared in nitric acid (5.0 mL, 65 %) from the original standard solution (1000.0 ppm) to the concentrations of 0.0 (blank), 0.1, 0.2, 0.5 and 1.0 ppm. The intensity of the rhodium was measured at the selected rhodium atomic wavelength. The limits of detection (LOD) were calculated using the calibration curve data (r2 = 0.9993) as three times the standard deviation (SDb)ofthe rhodium intensityobtained from 10 blankreplicates,divided by the gradient (m) of the calibration curve (sensitivity); LOD= 3 xSDb/m. The LOD was found to be 0.0041 ± 0.0003 ppm and the limit of quantitation (LOQ) was calculated as ten times the limit of detection and was found to be 0.041 ± 0.003ppm. The LOD (0.01005 ± 0.00002) and LOQ (0.1005 ± 0.0002) for the cobalt internal standard method were determined from the same formula with a different calibration curve. The rhodium intensities of the standards were plotted against the rhodium concentrations to give a calibration curve with a correlation coefficient (r2) of0.9996, a gradient (sensitivity) of0.2984 and an intercept of 0.0043.
6.2. Quantification of Rhodium in CRM
6.2.1. Dissolution of the CRM Samples using Microwave Digestion in Different Mineral Acids
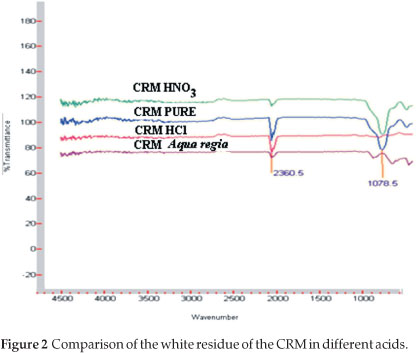
Eight dried CRM samples (~0.50 g) were accurately weighed (0.1 mg) andquantitativelytransferredintodifferentmicrowave PTFE vessels. To each pair of PTFE vessels, equal volumes of either hydrochloric, aqua regia, nitric or sulphuric acid (8 mL) were added and the samples digested under the microwave conditions specified in Table 2. The resultant mixtures were cooled, filtered and transferred into different volumetric flasks (100 mL). The samples were filled up to the mark, homogenized and left to stabilize for five hours before analyzed for rhodium content. The CRM residue together with the undigested starting material were analyzed using IR to determine if there were any changes before and after digestion as shown in Fig. 2. The rhodium recovery, which indicates the effectiveness of these acids in dissolving the CRM, is reported in Table 3.

6.2.2. Dissolution of the CRM Samples using Microwave Digestion in HCl
Five dried CRM samples (~0.50 g) were accurately weighed (0.1 mg) and quantitatively transferred into different microwave PTFE vessels. To each PTFE vessel, equal volumes of HCl (8 mL) were added and the samples digested under the microwave conditions specified in Table 2. The resultant mixtures were cooled, filtered and the filtrate heated to almost dryness. The chloride ions were removed from the CRM samples as chlorine gas with the periodic addition of nitric acid (two portions of 5 mL of 65 % HNO3) to limit the presence of EIE's at this stage of the investigation according to Equation 1. Nitric acid (5 mL, 65 %) was added to the dry precipitate and then quantitatively transferred to volumetric flasks (50.0 mL) and filled up to the mark using double-distilled water to yield a rhodium concentration of approximately 3.4 ppm depending on the weighed mass. The samples were homogenized and left to stabilize for five hours before analyzed for rhodium.
6.2.3. Preparation of Yttrium and Cobalt Internal Standard Solutions Equal volume and concentration of yttrium and cobalt internal standards (2.00 mL, 100.0 ppm) were pipetted into 100 mL volumetric flasks containing rhodium standard solutions with a working range of 0.5-10.0 ppm. Nitric acid (5.0 mL, 65 %) was added and the solutions were filled up to the mark using the double-distilled water and mixed thoroughly to obtain homogeneous solutions. The solutions were left to stabilize for five hours before use.
6.2.4. Preparation of the External Calibration Curve Calibration standards were prepared from the original standard solution (1000.0 ppm) to yield concentrations of 0.5, 1.0, 2.0, 5.0 and 10.0 ppm in different volumetric flasks. Nitric acid (5.0 mL, 65 %) was added and the flasks filled up to the mark using double-distilled water. The solutions were homogenized and allowed to stabilize for five hours before they were used. The intensity of the rhodium was measured at the selected atomic wavelength for rhodium at 343.489 nm. The rhodium intensities of the standards were plotted against the rhodium concentrations to give a calibration curve with a correlation coefficient (r2) of 0.9993, a gradient (sensitivity) of 0.735 and an intercept of 0.0011. The results for the rhodium determination in the CRM are tabulated in Table 3.
6.2.5. Preparation of the Standard Addition Calibration Curve
Five replicates of the CRM (~0.50 g) were prepared as outlined in Section 6.2.2 in HCl. Aliquots (10.0 mL) of the prepared stock solutions were pipetted into the prepared rhodium standard solution of concentrations 0.5, 1.0, 2.0, 0.5 and 10.0 ppm. Nitric acid (5.0 mL, 65 %) was added to each solution to yield a theoretical rhodium concentration of approximately 3.4 ppm. The rhodium intensities of the standards plotted against the rhodium concentrations gave a calibration curve with a correlation coefficient (r2) of 0.999(6), gradient of 7.62 and an intercept of 16.4. The results for the rhodium determination in the CRM are discussed in Section 7.3.
6.2.6. Preparation of the Internal Standard Addition Calibration Curves
Samples of ARM-504 (CRM) were prepared as described in Section 6.2.2. The same volume of the yttrium and cobalt stock solution (2.00 mL; 100.0 ppm) was also pipetted into all the rhodium CRM analyte solutions to match the concentration level of the internal standards. The prepared standard solutions and CRM samples were left to stabilize for five hours before analyzed for rhodium content. The intensity of the rhodium was measured at the selected atomic wavelength of, 343.489 nm, ionic wavelength for yttrium, 371.030 nm and ionic wavelength for cobalt, 228.616 nm. The ratio of rhodium to yttrium or cobalt intensities were plotted against the rhodium concentrations, which gave calibration curves with a correlation coefficient (r2) of 0.9999 for both, a gradient (sensitivity) of 0.0203 (Y), 0.2906 (Co) and an intercept of 0.0026 (Y), 0.0261 (Co).
6.3. Preparation of Rhodium Metal Powdered Samples and the Determination of Rhodium using the Direct Calibration and Cobalt Internal Standard Methods
Ten rhodium stock solutions were prepared (five each for every method) with accurately (0.1 mg) weighed powdered rhodium samples (~0.004 g) that were quantitatively transferred into microwave PTFE vessels. The samples were digested the same way as outlined in Section 6.2.2 in HCl. The resultant burgundy red solutions, with no precipitate, were then heated to almost dryness and nitric acid (5.0 mL, 65 %) was added to each sample prior to dryness and re-heated. The nitric acid addition process was repeated to facilitate the removal of most of the chloride ions (Equation 1) and to ensure proper acid matrix correlation of the analyte samples with those of the rhodium standard solutions. Finally, nitric acid (5 mL, 65 %) was added to the almost dry precipitates and then quantitatively transferred to volumetric flasks (100.0 mL) while the cobalt standard was added (2.0 mL; 100.0 ppm) to the set of standards that were used for internal standard addition analysis. The analyte volumetric flasks were filled to the mark using double-distilled water to yield a theoretical rhodium concentration of approximately 3.4 ppm. The analyte solutions were homogenized and left to stabilize for at least five hours before they were analyzed for rhodium content using (i) the direct calibration method and (ii) the cobalt internal standard.
6.4. Preparation of theRhCl3·3H2O Solutions
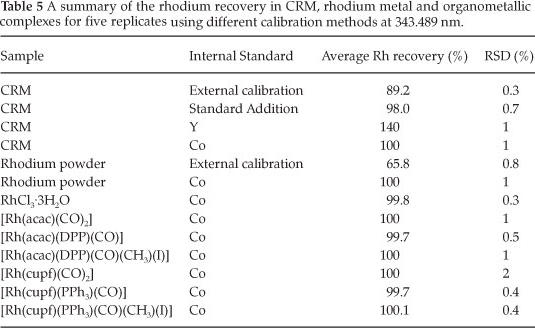
The burgundy red crystals of RhCl3·3H2O (~0.05 g) were accurately weighed (0.1 mg) and dissolved in a volumetric flask (100.0 mL). Subsequent dilutions were made to prepare a solution of ~3 ppm. To each of the analyte samples, aliquots of the cobalt internal standard (2.00 mL; 100.0 ppm) and nitric acid (5.0 mL; 65 %) was added. The volumetric flasks were filled to the mark using double-distilled water and left to stand for five hours to stabilize before analysis. The samples were quantified for rhodium content against the rhodium/cobalt calibration curve with a correlation coefficient (r2) of 1.00, gradient of 0.2403 and an intercept of 0.0113. The results are reported in Table 5.
6.5. Preparation of the Organometallic Complexes for Rhodium Determination using the Cobalt Internal Standard
Samples of different rhodium organometallic complexes ([Rh(acac)(CO)2],42 [Rh(acac)(DPP)(CO)], [Rh(acac)(CO)(DPP) (Me)(I)],43 [Rh(cupf)(CO)2],44 [Rh(cupf)(PPh3)(CO)]45 and [Rh(cupf)(PPh3)(CO)(Me)(I)]46 where synthesized and characterized by IR spectra. Samples of these complexes were accurately weighed and quantitatively transferred to 100.0 mL beakers. Hydrochloric acid (5 mL; 32 %) was added and the samples were digested (ca. 110 ° C) in open beakers until the samples were completely dissolved. The dissolved samples were further heated at the same temperature to almost dryness and nitric acid was added (5 mL; 65 %) prior to dryness and the solution re-heated. The addition of nitric acid to the almost dry samples was repeated three times before the samples were quantitatively transferred into 100.0 mL volumetric flasks. Nitric acid (5.0 mL; 65 %) was added to the stock solutions and filled to the mark using double-distilled water. Different aliquots of the sample stock solutions were pipetted into volumetric flasks (100.0 mL) to yield a rhodium concentration of approximately 3.40 ppm. Cobalt internal standard (2.0 mL; 100.0 ppm) and nitric acid (5.0 mL; 65 %) was added into each analyte solution. The analyte flasks were filled to the mark using double-distilled water, homogenized and left to stabilize for five hours before rhodium content analysis. The samples were quantified for rhodium content against the rhodium/cobalt calibration curve with a correlation coefficient (r2) of 1.00, gradient of 0.2319 and an intercept of 0.0136 and the results are reported in Table 5.
6.6. The Effect of Unmatched Matrixes Towards Rhodium Recovery using the Cobalt Internal Standard
6.6.1. Preparation of RhCI3·3H2O Solutions in Different Acids (HCl, HBr and HNO3) and the Quantitative Determination of Rhodium
Aliquots of RhCl3·3H2O, as prepared in Section 6.4, were pipetted into separate volumetric flasks (100.0 mL) to yield a rhodium concentration of ca. 3.4 ppm. Increasing amounts (0-0.12 moles) of HCl, HBr and HNO3 as shown in Fig. 4 were added to each analyte solution. Cobalt internal standard (2.00 mL; 100.0 ppm) was added and the volumetric flasks were filled to the mark using distilled water and homogenized before left to stand for five hours. The samples were quantified for rhodium content with the rhodium/cobalt calibration standards prepared in Section 6.4 and the results are given in Fig. 3.

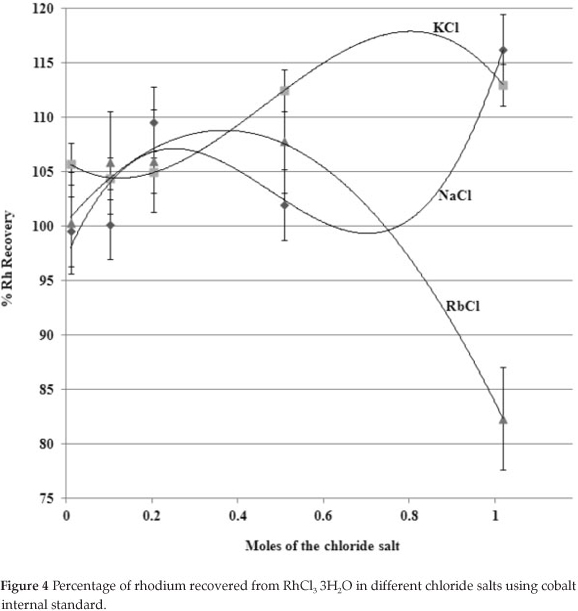
6.6.2. Preparation of the RhCl3·3H2O Solutions and the Presence of Different Halide Salts
Different aliquots of the prepared 10.0 M stock solutions of NaCl, KCl and RbCl were pipetted in increasing order to yield 0.01, 0.10, 0.20, 0.50 and 1.00 moles of the salt solution. RhCl3·3H2O prepared in Section 6.4 was spiked in each volumetric flask to yield a rhodium concentration of ca. 3.4 ppm. Cobalt internal standard (2.0 mL; 100.0 ppm) and nitric acid (5.0 mL, 65 %) was added to each flask and filled with double-distilled water. The solutions were homogenized and left for five hours to stabilize before rhodium content analysis using the (i) direct calibration curve and (ii) a cobalt internal standard. The samples were quantified for rhodium content with the rhodium calibration standards prepared in Section 6.4 and the results are given in Fig. 4.
7. Results and Discussion
7.1. Wavelength Selection and the Determination of Detection and Quantification Limits of Rhodium
Qualitative analysis of the CRM was carried out to determine the composition of the elements present in the CRM in order to select the most appropriate wavelength with a minimum interference for rhodium analysis. From the qualitative analysis, nineteen different elements were found to be present and these elements included the EIEs; Na, Li,Mg, Ca, Rb, Ba and Cs, transition metals; Al ,Ti, V, Ga, Sr,Ti and Pb and the so-called base metals; Cr, Fe, Ni, Cu and Zn. These three groups cause spectral interference by either enhancing or suppressing the analyte signal during the quantitative determination of PGM.36,47-49 A quantitative analysis was carried out to determine the concentrations of the EIEs, transition and base metals in the CRM to establish an appropriate method, conditions and a suitable internal standard element for the rhodium analysis. The concentration of EIEs was less than 2 ppm while concentrations of between 5-1000 ppm were obtained for the transition and base metals. The results from the ICP-OES 'profile' function (Fig. 1) identified the wavelength at 343.489 nm as the most appropriate line for the accurate quantitative analysis of rhodium in the presence of all the transition and base metals, while the low concentration of the EIE's promised very little spectral influence in rhodium quantification.
The experimentally determined LOD/LOQ for both the direct calibration and the cobalt internal standard methods of 0.00408/0.0408 ppm and 0.01005/0.1005 respectively, points to the opportunity of accurate determination of rhodium at very low concentrations using ICP-OES as an analytical tool. This is indicated by the small differences in the LOD and LOQ (0.00597 and 0.0597 respectively) of these methods.
7.2. Microwave Dissolution of the CRM Samples and the Quantitative Determination of Rhodium using Direct Calibration
Initial analytical results, as well as previous studies50 indicated that rhodium analysis is extremely sensitive to matrix matching or the absence of it. In that respect care was taken from the start of this study to ensure proper matrix matching by firstly removing excess HCl (which turned out to be the preferred acid for CRM dissolution as discussed in the next paragraph) and secondly by the addition of identical volumes of HNO3 to the analyte samples and the standard solutions. The first few additions of nitric acid (see Paragraph 6.2.2) to the reaction mixture containing the CRM samples ensured the removal of the chloride ions via the formation of the volatile nitrosyl chloride and chlorine gas products according to Equation 1.
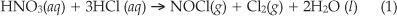
The formation of the nitrosyl chloride and chlorine was observed by the fuming nature of the reaction and the characteristic yellow colour of chlorine gas, which was formed during the reaction.
Interesting results were obtained for the microwave-assisted dissolution of the grey-brown CRM in the presence of different acids (see Table 3). These quantitative results (external calibration) clearly indicated a higher rhodium recovery with HCl (89 %) as dissolution medium compared to aqua regia (80 %), which has widely been used as the preferred dissolution medium of PGM. Even lower recoveries for HNO3 (44 %) and H2SO4 (33 %) were recorded. The lower than expected recovery of the rhodium was initially attributed to the incomplete digestion of the samples using this digestion technique or its prevailing reaction conditions as was evidenced by the precipitate (white - HCl; whitesh-brown - aqua regia; yellowish-brown -HNO3; greyish-brown - H2SO4) that remained after the two-hour digestion period (Table 2).The duration of the digestion of the CRM was extended for another hour under the same robust conditions. No visual improvement in the results (less remaining precipitate or total disappearance of the precipitate) or percentage rhodium recovery was obtained with this change in experimental condition. Infrared spectra of the different precipitates obtained from the acid dissolution reactions were compared with that of the original CRM as shown in Fig. 2. The correlation between the decrease in the stretching frequency at 1078.5 cm-1 from HNO3 to HCl (complete disappearance) with a steady increase in rhodium recovery from HNO3 to HCl suggested the complete rhodium dissolution/leaching from the CRM in the presence of HCl.
The combination of the complete disappearance of the IR peak at 1078 cm-1 with the relatively high rhodium recovery with HCl as dissolution medium motivated a change in calibration method so that a suitable alternative for external calibration was found.
7.3. Quantitative Determination of Rhodium in the CRM using Standard Addition Calibration
In an attempt to optimise the percentage rhodium recovery and to limit any possible interference due to the complicated sample matrix, standard addition calibration was initially used to quantify rhodium in the CRM. Results of rhodium recovery using this calibration method increased the average percentage recovery to 98.0 ± 0.7 % for five new CRM samples that were microwave digested in HCl. The quantification of rhodium in the CRM using this method was better than the direct calibration method (ca. 96.0 % compared to 88.0 %), but still not within the acceptable analysis recovery range due to the possibilities of matrix interference. The major drawback of this method was that it required a larger volume of both the standard and analyte solutions, which had to be prepared for every successive analysis. The internal standard addition as calibration method was subsequently used in an attempt to eliminate either the solubility
7.4. Internal Standard Selection and Rhodium Quantification in the CRM using Yttrium and Cobalt as Internal Standards
The selection of the appropriate internal standard was very important in the accurate determination of rhodium. The element of choice is critical because the recovery of the element is highly dependent on the choice of the internal standard. According to the literature,51-53 yttrium is the most preferred and widely used internal standard for the determination of PGM. Several ISO approved methods use yttrium as internal standard for PGM quantification.54-56 Elements like tin, indium, copper, nickel, silver, thallium, molybdenum and lead have been used as internal standards in different spectrometric techniques such as ICP-MS as discussed by Salin et al.57
Cobalt and rhodium are in the same group (9 or VIIIb) on the periodic table and have similar 1st, 2nd and 3rd ionization energies (Table 4), but are significantly different from those of yttrium. The similarity of ionization energies between cobalt and rhodium led us to believe that cobalt can be used as a possible internal standard for rhodium analysis. The ionic lines for yttrium and cobalt at 371.030 and 228.616 nm respectively, were chosen as the lines for analysis.58
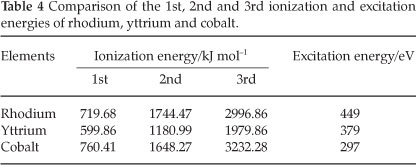
Recoveries in excess of 100 % were obtained for all five CRM samples with an average of 140(1) % using yttrium as internal standard. The positive error in rhodium recovery with the use of yttrium as internal standard points to interference at the selected wavelength for rhodium analysis.36,20 Excellent rhodium recoveries of ±100(1) % were obtained using cobalt as internal standard (see Table 5). The successful recovery of the rhodium in the CRM samples also confirms the successful dissolution/leaching of rhodium from the CRM using HCl as dissolution medium.
The success of the cobalt as internal standard is attributed to its ability to behave in a similar manner as the rhodium analyte within the ICP flame. The use of cobalt as internal standard was therefore chosen as the preferred method for rhodium analysis in the rest of this study on the basis of excellent recovery and its ability to accurately compensate for the matrix-induced signal variations as well as being specific. This method of rhodium quantification was subsequently evaluated on rhodium metal powder of high purity (RM).
7.5. Quantitative Determination of Rhodium from Powdered Rhodium (99.9 %)
Rhodium metal (99.9 % purity) was firstly analyzed using the direct or external calibration method due to the simplicity of the sample matrix and secondly using cobalt as internal standard. The quantitative results obtained from the experimental analysis using cobalt as internal standard (Table 5) revealed excellent rhodium recovery of 100(1) % compared to an average of 65.8(0.8) %, which was obtained from the direct calibration method. The direct calibration method clearly indicated, as was the case for the CRM, that this method was not suitable for the accurate determination of rhodium. A qualitative study indicated the presence of K, Na, Mg, Ca, Fe, Pd, Ag and Pt at micro and ultra-micro level. The excellent rhodium recovery also confirmed the quantitatively dissolution of the rhodium metal withacid-assisted microwave digestioninthe presence ofHCl.
7.6. Quantitative Determination (Method Validation) of Rhodium in Different Inorganic and Organometallic Samples using Cobalt as Internal Standard
The use of cobalt as internal standard was validated using other samples which contained rhodium at different percentage levels. These samples included the water soluble inorganic RhCl3∙3H2O salt (amount of water molecules verified with TG and DSC) and a number of rhodium containing organometallic complexes such as [Rh(acac)(CO)2] and [Rh(cupf)(PPh3) (CO)(CH3)(I)]. Organometallic complexes were selected on the basis of experience and ease of preparation, but more importantly on the purity of the products. In many cases the products had been characterized by means of X-ray crystal structure determinations, which confirmed the chemical formula and therefore the rhodium content.
Excellent rhodium recoveries in excess of 99 % (see Table 5) were obtained for all the samples that were selected or synthesized. Thus, the use of cobalt as internal standard for the successful recovery of rhodium in a variety of chemical compounds on condition that matrix matching is ensured, was validated.
7.7. The Effect of Unmatched Matrixes Towards Rhodium Recovery using the Cobalt Internal Standard
The effect of the group (I) EIE's on rhodium recovery in RhCl3∙3H2O using external calibration and cobalt internal standard calibration (robustness of the method) was investigated. Elements were added as the respective chloride salts into the final analyte mixture. Amounts were increased in a stepwise fashion and were identical for each solution in order to isolate the cation effect on rhodium recovery. In the event that Cl- ions interfered with the analysis, the effect should have been the same in all the analyte solutions at the different concentration levels. The effect of EIE's on rhodium recovery is shown in Figs. 3 and 4. An initial rhodium recovery of lower than 100 % with external calibration is seen in Fig. 3. Initially, the addition of halide salts (an increase in metal chlorides) was accompanied by a similar decrease in rhodium recovery. This suggested that the influence of the chloride ion addition overshadowed the possible influence of metal ions on the rhodium recovery. A drastic change in rhodium recovery took place from about 0.1 moles indicating an increased influence of the metal ion. The most profound effect was caused by RbCl with a constant decrease in rhodium recovery while the effect of NaCl was the smallest. A totally different effect was observed when cobalt was used as internal standard (Fig. 4). In this case all the rhodium recoveries started at 100 % and then increased to about 107 % at 0.2 moles for all the EIE. This also suggested that the influence of chloride ions override the metal ion influence. The influence of the EIEs changed after this point with either an increase in rhodium recovery with NaCl addition or a decrease with RbCl addition. Thus, there was an increase in metal ion influence on rhodium recovery at higher EIE concentrations. The same effect of sodium ions on the quantitative determination of PGM was observed by De Klerk et al.59
The isolation of the anion influence from the acid effect is shown in Fig. 5. The initial rhodium recovery started at 100 % with prevailing matrix matching conditions. The addition of extra acid decreased initial rhodium recovery for all three acids suggesting a common acid effect (acid amount the same for all three acids). On further acid addition, however, the graphs changed substantially for the different acids, suggesting that the anion over-rode the rhodium recovery. The largest effect was observed with the addition of HBr.
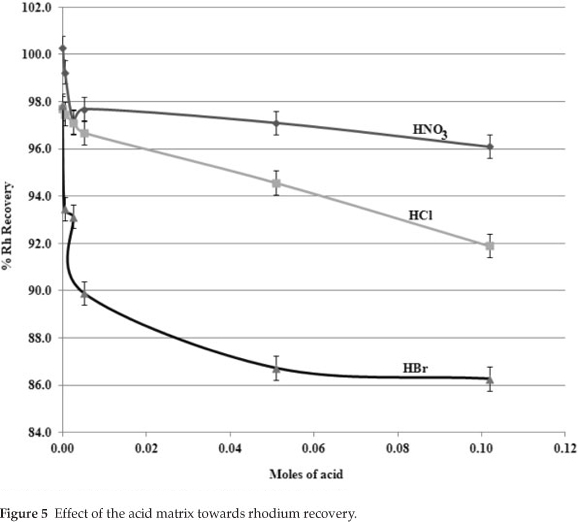
These results show that both anions and cations (EIE) have a pronounced effect on rhodium recovery using the new analytical method. Thus, the accurate recovery of rhodium depends heavily on the proper matrix matching.
8. Validation Parameters
Total recovery was obtained for all the samples (z-test for CRM and t-test for all the other samples at a 95 % confidence interval) with relative standard deviations (RSD) between 0.26 and 1.87 for all the samples.60 The regression coefficient of all the calibration ranged from 0.9997 to 1.00, standard deviation of the slope (sa) ranged from 0.00029 to 0.00936, and the intercept (sb) and was between 0.00102 and 0.03338. Intercepts of between 0.0136 and 0.0261 (blank solution) for all the samples confirmed the selective analysis of rhodium at 343.489 nm. The sensitivity of the rhodium recovery with a slight change in the total acid concentration in the samples rendered the method not robust. The rhodium limit of detection (LOD) and limit of quantification (LOQ) were determined to be 0.01005 ± 0.00002 and 0.1005 ± 0.0002 ppm respectively, which is feasible for measuring trace amounts of rhodium.
9. Conclusion
A matrix matching protocol accompanied by the cobalt internal standard circumvented the matrix problem by compensating for the dominant matrix components caused by EIEs, transitional metals, base metals, background emission and other fluctuations occurring during the sample introduction and measurement. Excellent results in rhodium recovery were obtained using cobalt internal standard as its emission intensity showed no interference at the second most intense line, 228.616 nm. As yttrium is commonly considered as a useful internal standard for all the PGM determination,53 its signal instability in the presence of other metals as detected in the CRM rendered it less suitable for rhodium determination as demonstrated by the high rhodium recoveries. The results of the cobalt internal standard showed to be in total agreement with the certified values of the CRM. The subsequent determinations of rhodium from different samples such as the rhodium metal, RhCl3∙3H2O and organometallic complexes were acceptable at 95 % confidence intervals. The method was found to be less effective in an unmatched matrix and the results obtained were in agreement with those obtained by Garden et at.61 who reported that ICP-OES signals can be suppressed by up to 40 % if the acid matrices are not matched. The acid matrix match between the standards and the analyte samples were therefore vital for the correct and accurate recovery of rhodium in different types of samples.
Acknowledgements
The authors thank the Research Fund of the University of the Free State, the National Research Foundation of South Africa and Inkaba yeAfrica for financial support.
References
1 http://www.kitco.corn/charts/ (accessed on 24/04/12). [ Links ]
2 G. Von Gruenenwaldt, Miner. Sci. Engng., 1977, 9(2), 83-96. [ Links ]
3 S.S. Cramer and J.C. Bernard, Materials Park, ASM International, 1990, pp. 393-396. [ Links ]
4 D.R.Lide, CRC Handbook of Chemistry and Physics: a Ready-reference Book of Chemical and Physical Data. CRC Press, Boca Raton, 2004, pp. 4-26. [ Links ]
5 http://www.britannica.com/EBchecked/topic/501671/rhodium (cited on 17/09/09). [ Links ]
6 R. Jana and J.A. Tunge, Org. Lett, 2009, 11(4), 971-974. [ Links ]
7 D.A. Evans and M.M.Morrissey, J. Am. Chem. Soc., 1984, 106(13), 3866–3868. [ Links ]
8 W.S. Knowles and M.J. Sabacky, Chem. Commun., 1968, 22, 1445-1446. [ Links ]
9 W.S. Knowles, J. Chem. Educ, 1986, 63(3), 222-225. [ Links ]
10 H.Y.H. Gao and R.J. Angelici, Organometallics, 2000, 19(4), 622-629. [ Links ]
11 R. Cramer, J. Am. Chem. Soc., 1967, 89, 1633-1639. [ Links ]
12 R.R. Brooks, The analytical chemistry of the noble metals, in Noble Metals and Biological Systems: Their Role in Medicine, Mineral Exploration, and the Environment, (R.R. Brooks, ed.), CRC Press, Boca Raton, FL, 1992, pp. 17-20. [ Links ]
13 S. Calmotti, C. Dossi, S. Rechia and G.M. Zanderighi, Spectrosc. Eur., 1996, 8, 18-22. [ Links ]
14 R.R. Barefoot, Anal. Chim. Acta, 2004, 509, 119-125. [ Links ]
15 M. Balcerzak, Anal. Sci, 2002, 18, 737-750. [ Links ]
16 F.E. Beamish, The Analytical Chemistry ofthe Noble Metals, Pergamon Press, Oxford, 1966. [ Links ]
17 A. Marucco, Nucl. Instr. Meth. Phys. Res., 2004, B213, 486-490. [ Links ]
18 S. Greenfield, H. McD. McGeachin and P.B. Smith, Anal. Chim. Acta, 1976, 84, 67-78. [ Links ]
19 P.WJ.M. Boumans, Opt. Pura. Apl., 1978, 11, 143-171. [ Links ]
20 P. Schramel and J. Ovcar-Pavlu, Fresenius Z. Anal. Chem., 1979, 298, 28-31. [ Links ]
21 S. Xi-En and C. Qi-Lan, Spectrochim. Acta, 1983, 38B, 115-121. [ Links ]
22 W.Z. Krasil'shchik, E.I. Voropaev, E.J. Shmakova and M.S. Shchupakhin, Vysokoch Veshchestva, 1987, 1, 151-157. [ Links ]
23 R.L. Dahlquist and J.W Knoll, Appl. Spectrosc, 1978, 32, 1-30. [ Links ]
24 J.L. Imbert and J.M. Mermet, Analysis., 1984, 4, 209-219. [ Links ]
25 H. Kawaguchi, T. Ito and A. Mizuike, Spectrochim Acta, 1980, 35B, 199. [ Links ]
26 V.P. Baluda, L.N. Filimonov, V.G. Miskar'yants and V.V. Nedler, in RF-ICP/AES: Sborniknauchntrudov, (H.I. Zil'bershtein, ed.), Nauka, Moscow, 1987, pp. 75-93. [ Links ]
27 K. Boch, M. Schuster, G. Risse and M. Schwarzer, Anal. Chim. Acta, 2002, 459, 257-265. [ Links ]
28 O.V Borisov, D.M. Coleman, K.A. Oudsema and R.O. Carter III, J. Anal. At. Spectrom., 1997, 12, 239-246. [ Links ]
29 I.V. Kubrakova, T.F. Kudinova, N.M. Kuz'min, I.A. Kovalev, G.I. Tsysin and Y.A. Zolotov, Anal. Chim. Acta, 1996, 334, 167-175. [ Links ]
30 N.S. Mokgalaka, R.I. McCrindle, B.M. Botha and L. Marjanovic, S. Afr. J. Chem., 2002, 55, 72-86. [ Links ]
31 R.I. Botto, Spectrochim Acta, 1985, 40B, 397-412. [ Links ]
32 A. Delijska and M. Vouchkov, Fresenius Z. Anal. Chem., 1985, 321, 448-452. [ Links ]
33 J-L. Todolí, S. Maestre, J. Mora, A. Canals and V. Hernandis, Fresenius J. Anal. Chem., 2000, 368, 773-779. [ Links ]
34 K.W. Jackson and S. Lu, Anal. Chem., 1998, 70, 363-384. [ Links ]
35 R. Jenkins, X-Ray Fluorescence Spectrometry, John Wiley & Sons, New York, 1988, pp. 465-487. [ Links ]
36 G.J. Schmidt and W. Slavin, Anal. Chem., 1982, 54, 2491-2495. [ Links ]
37 Z. Zadgorska, H. Nickel, M. Mazurkiewicz and G. Wolff, Fresenius Z. Anal. Chem., 1983, 314, 356-361. [ Links ]
38 www.finishing.com/445/01.shtml (accessed 30 Aug 2010). [ Links ]
39 S.S. Basson, J.G. Leipoldt and J.T. Nel, Inorg. Chim. Acta, 1984, 84 167-172. [ Links ]
40 S.S. Basson, Mikroanalytisches Labor Pascher HPT 1 and HPT 2, 1986. [ Links ]
41 J.A. Bailey, S.L. Grundy and S.R. Stobart, Inorg. Chim. Acta, 1996, 243, 47-56. [ Links ]
42 J.G. Leipoldt, S.S. Basson, L.D.C. Box and T.I.A. Gerber, Inorg. Chim. Acta, 1978, 26, L35-L37. [ Links ]
43 S.S. Basson, J.G. Leipoldt, A. Roodt, J.A. Venter and T.J. Van der Walt, Inorg. Chim. Acta, 1986, 119, 35-38. [ Links ]
44 S.S. Basson, J.G. Leipoldt, A. Roodt and J.A. Venter, Inorg. Chim. Acta, 1986, 118, L45-L47. [ Links ]
45 J.A. Venter, Structural and Kinetic Study of Rhodium Complexes of N-aryl-N-nitrosohydroxylamines and Related Complexes, Ph.D. thesis, University of the Free State, Bloemfontein, South Africa, 2006, p.105. [ Links ]
46 M.P. Coetzee, Characterization and Oxidative Addition of Different Rhodium(I) Carbonyl Diphenyl-2-pyridylphosphine Complexes, M.Sc. thesis, University of the Free State, Bloemfontein, South Africa, 2008, pp. 57-58. [ Links ]
47 M.W. Blades and G. Horlick, Spectrochim. Acta, Part B., 1981, 36, 881-900. [ Links ]
48 X. Jian, L Qingyuan, L. Wenchong, Q. Haowen, T. Jingyuan and Z. Zhanxia, J. Anal. At. Spectrom., 1992, 7, 131-134. [ Links ]
49 G.F. Larson and VA. Fassel, Anal. Chem., 1976, 48, 1161-1166. [ Links ]
50 E.G. Chudinov, I.I. Ostroukhova and G.V Varvanina, Fresenius Z. Anal. Chem., 1989, 335, 25-33. [ Links ]
51 G.A. Zachariadis and P.C. Sarafidou, Microchim. Acta, 2009, 166, 77-81. [ Links ]
52 J.C. Ivaldi and J.F. Tyson, Spectrochim. Acta Part B, 1996, 51, 1443-1450. [ Links ]
53 R. Kovacevic, M. Todorovic, D. Manojlovic and J. Mutic, J. Iran. Chem. Soc, 2008, 5, 336-341. [ Links ]
54 International Organization for Standardization: Determination of Palladium in Palladium Jewellery Alloys - Method Using Inductively Coupled Plasma Emission Spectrometry on a Solution with Yttrium as Internal Standard. Doc. ISO/DIS 11495.2. [ Links ]
55 International Organization for Standardization: Atomic Absorption Doc. ISO/WD 11492. [ Links ]
56 International Organization for Standardization: Determination of Platinum in Platinum Jewellery Alloys - Method Using Inductively Coupled Plasma Emission Spectrometry on a Solution with Yttrium as Internal Standard. Doc. ISO/DIS 11494.2. [ Links ]
57 E.D. Salin, M. Antler and G. Bort, J. Anal. At. Spectrom., 2004, 19, 1498-1500. [ Links ]
58 T.T Chiweshe, Quantification of Rhodium in Series of Inorganic and Organometallic Compounds, M.Sc. thesis, University of the Free State, Bloemfontein, South Africa, 2010, pp. 97-99. [ Links ]
59 http://physics.nist.gov/PhysRefData/IonEnergy/tblNew.html (accessed 17 September 2010). [ Links ]
60 A. De Klerk and C. J. Rademeyer, J. Anal. At .Spectrom., 1997, 12, 1221-1223. [ Links ]
61 D.A. Skoog, F.J. Holler and T.A. Nieman, Principles of Instrumental Analysis, 5thedn, Saunders College Publishing, Philadelphia, 1998, pp. 149-153. [ Links ]
62 L.M. Garden, J. Marshall and D. Littlejohn, J. Anal. At. Spectrom., 1991, 6, 159-163. [ Links ]
Received 12 June 2012
Revised 28 October 2012
Accepted 23 November 2012
* To whom correspondence should be addressed. E-mail: purcellw@ufs.ac.za

