










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
RESEARCH ARTICLE
Optimization of experimental parameters in preparing multinanoporous TiO2 thin films by the anodic oxidation method
Gracien B. Ekoko*; Joseph K-K. Lobo; Omer M. Mvele; Antoine K. Mbongo; Jérémie L. Muswema*; Clarisse Z. Lefuni
Department of Chemistry, University of Kinshasa, B.P. 190, Kinshasa XI, Democratic Republic of Congo
ABSTRACT
The anodic oxidation method has been applied to the preparation of multinanoporous TiO2 thin films. The experimental parameters, including the electrolyte nature, oxidation voltage, and oxidation time have been carefully controlled. Their influence on the structure, morphology and photocatalytic activity of the prepared TiO2 films has been evaluated by measuring the current density. The result showed that there was a relatively wide range of preparation conditions, and the internal relationship between the structure and the photocatalytic properties of the TiO2 films was analyzed.
Keywords: Titanium films, anodic oxidation, multinanoporous films, photocatalytic activity.
1. Introduction
In the most recent research devoted to water purification, TiO2-mediated photolytic catalysis is considered as an advanced oxidation process, which produces highly degrading hydroxyl radicals (HO●). These radicals generated in solution are strong oxidative species, responsible for the oxidation of organic pollutants present in waste water.1-6
When one molecule of the TiO2 catalyst (band gap 3.2 eV) is exposed to light whose energy exceeds the energy of the band gap of the semiconductor, an electron (e-) is promoted from the valence band (VB) to the conduction band (CB), leavingbehind a hole (h+) inthevalenceband.Therefore,aportionofthe photo-excited electron-hole pairs diffuse onto the surface of the catalyst. The charge-carriers (e- and h+) produced are trapped at the surface of the semiconductor and take part in the chemical reaction. The holes can oxidize organic pollutants present in water, since they can react with water in a one-electron oxidation step to produce the highly reactive hydroxyl radical (HO●).
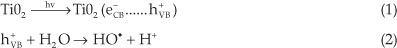
The use of TiO2 in wastewater treatment is limited, due to its low energy conversion efficiency of less than 5 %. Indeed, the majority of the photogenerated charge carriers (e- and h+) undergo recombination before they reach the surface of TiO2 where they can interact with the adsorbed molecules. Because of this recombination, up to 90 % of the generated carriers are lost within 10-9 s after their production. The percentage of charge carrier recombination thus has a major effect on the photo-catalytic efficiency of TiO2. It was also reported that as the grain size of TiO2 decreases, the probability of charge carrier recombination also decreases, thus resulting in an improvement of the TiO2 photocatalytic properties.7
Furthermore, the use of porous materials has proved advantageous in this respect. Indeed, well-defined porous structures have a higher surface area which offers a special environment for the degradation of the organic pollutants. Additionally, materials with porous morphology allow faster diffusion of reactive oxygen species, and in this case, the organic substrates are adsorbed within the catalyst surface where they are efficiently degraded.8-9
For the application of TiO2 in the field of wastewater detoxification, the semiconductor photocatalyst powder is usually dispersed in a liquid suspension. After the degradation process under UV light, the TiO2 powder remaining suspended in the water has to be removed by centrifugation or by filtration, and this is costly.
By contrast, a TiO2-fixed-system needs no such operation and continuous wastewater treatment becomes more practical. Several investigators have dealt with the development of procedures for catalyst immobilization on a substrate (solid support). TiO2 molecules have been fixed to a variety of surfaces: glass,10 silicon,11 clays,12 organic polymers,13 thin films,14 concrete,15 alumina,16 and carbon.17 However, in most of these methods, the desired TiO2 electrodes were fabricated by pasting a slurry of TiO2 on a substrate, drying the pasted slurry for a time long enough to remove substantially all of the liquid and to stabilize the mechanical binder, thereby providing a TiO2 structure.
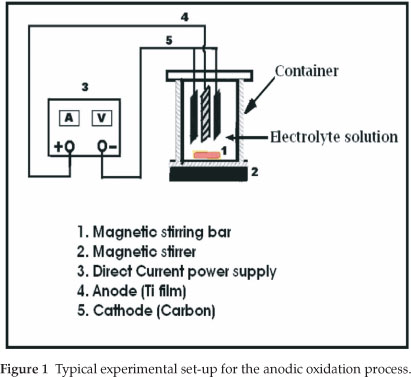
In the present work, a titanium (Ti) metal sheet is used as the starting material to produce titanium dioxide on the surface of Ti metal by anodic oxidation. The process, also called 'micro arc oxidation' (MAO), produces the anodic titania film with a good adhesion with the titanium substrate, higher specific surface area, and good chemical stability, which enable its application as a photocatalyst for the degradation of organic pollutants in waste water purification. Therefore, the TiO2 produced is immobilized on a Ti metal substrate. For this purpose, the Ti sheet is made the anode by connecting it to the positive terminal of a direct current (DC) power supply. The cathode consists of a carbon rod, because carbon is inert in the anodizing bath. At a constant anodization current, when a voltage is applied, the titanium substrate surface is expected to react with water and form an oxide layer on the titanium metal surface.
The aim of this paper was to select the best experimental conditions for producing multinanoporous TiO2 thin films (anatase) with a higher pore density, smaller TiO2 size and suitable photocatalytic properties. The multinanoporous morphology is needed because the pores could act as adsorption centres and so help improve the photocatalytic activity.18
2. Experimental
To prepare TiO2 thin films by anodic oxidation, H2SO4 and NaOH were selected as electrolytes. Titanium substrates were prepared from titanium plates of 0.5 mm thickness containing Ti (>97 %), N (<0.012 %), C (<0.02 %), Si (<0.04 %), and Fe (<0.06 %). They were cut into pieces of dimensions 7.5 × 1 cm2 and were cleaned in chloroform and etched in hydrofluoric acid (4 % w/w) for a period from 0.5 to 1 min, depending on the required surface structure. They were rinsed in distilled water, then in acetone before being dried and analyzed.19-20
Electrodes of TiO2 were prepared by anodic oxidation at room temperature by varying different parameters as listed in Table 1 where samples were labelled depending on the fixed experimental conditions. The current density was fixed to about 15-20 mA cm-2 for the preparation of these electrodes. The direct current (DC) power supply (150 V - 5 A) was used to adjust the anodization voltage, under a potentiostatic regime. Sulfuric acid H2SO4 (or sodium hydroxide NaOH) of different concentrations was used as electrolyte. Three different values of oxidation voltages were chosen (100,130 and 150 V). The H2SO4 concentrations selected were 0.5, 1.0 and 2.0 mol dm-3. The anodization time was varied from 5 to 11 min. After anodization, the products obtained were characterized with different analytical methods.

Since anodization is an exothermic process which leads to electrolyte heating, the solution was magnetically stirred during the oxidation process in order to maintain the temperature constant and thereby prevent local heating of the produced oxide film. Moreover, stirring the solution affects the rate at which electro-active molecules in the bulk solution are brought to the electrode surface by mass transfer. The experimental setup for the anodic oxidation is illustrated in Fig. 1.
To quantify the effect of the electrolyte nature in the production of TiO2, another experiment was designed at room temperature in alkaline electrolyte solution (NaOH) at two different concentrations (0.1 and 0.2 mol dm-3). The oxidation voltage was fixed at 130 V and the anodization time to 5 min. In the same way, acidic electrolyte solutions of H2SO4 with respective concentrations of 0.1,0.2,0.5,0.8 and 1.0 mol dm-3 were used at the same anodizing conditions to compare the efficiency of the prepared TiO2 electrodes. The experimental conditions are listed in Table 2.

The photocatalytic properties were investigated by using 0.2 mol dm-3 methanol solution as an organic pollutant model in the presence of sodium sulfate (0.3 mol dm-3) as a supporting electrolyte because of its high conductivity which can be translated into little or almost no ohmic resistance between the reference electrode and the working electrode so that the measured current can be considered as due exclusively to the diffusion of the CH3OH solution at the electrode surface.21
The Pyrex cell containing the CH3OH solution was externally irradiated with a 125 W high pressure mercury UV lamp (model GGZ-125, λ = 365 nm) provided by 'Shanghai Yaming Lamps Factory'. A cooling fan was used to prevent overheating of the solution in the Pyrex cell. The system was placed over the designed compartment covered by an aluminium foil.
All photoelectrochemical experiments were performed by using a computer-controlled EG&G Princeton Applied Research (PAR, model 273A) potentiostat. For this purpose, three electrodes were required for the measurement: the working electrode made of a TiO2 film, the counter electrode made of a platinum sheet (Pt) and a stable reference electrode (the saturated calomel electrode, SCE). All potentials were quoted with respect to the SCE. The measurement was made with CH3OH (0.2 mol dm-3) and H2SO4 (0.3 mol dm-3). The photocurrent densities were measured first in the dark and then under UV illumination. The potentiostat was used to fix a constant applied bias of 0.5 V, and the current densities generated were recorded.
3. Results and Discussion
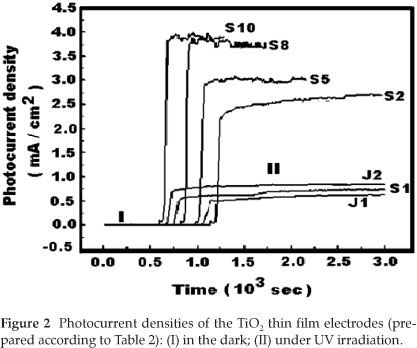
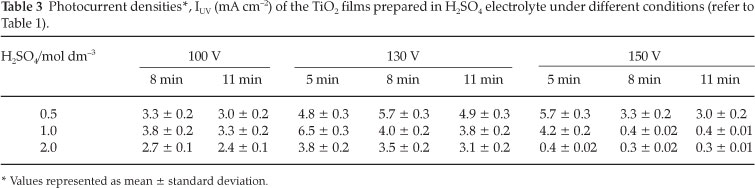
The photocurrent densities obtained with only H2SO4 as electrolyte, and when using NaOH and H2SO4 as electrolytes are respectively listed in Table 3 and Fig. 2 respectively.
From Table 3, it can be seen that the electrodes prepared at 130 V exhibited higher values of photocurrent densities compared to those obtained with the electrodes prepared at 100 V and 150 V The values of current densities for electrodes prepared at 130 V varied in the range between 3.1 mA cm-2 and 6.5 mA cm-2. The maximum value of the current density measured is designated IUV. By fixing the anodization time to 5 min and the H2SO4 electrolyte concentration to 1.0 mol dm-3, the photocurrent obtained for sample E5 was the highest and a value of IUV = 6.5 mA cm-2 was obtained.
From Fig. 2 and Table 2, one can observe that the films of TiO2 prepared in alkaline solutions (J1 and J2) exhibited smaller values of the current density (less than 1.0 mA cm-2). However, for the series of TiO2 films prepared in acid electrolyte (S1,S2,S5,S8 and S10), a significant increase in current density was observed.
This result indicates that the photoelectrochemical properties of TiO2 prepared in acid electrolyte depend significantly on the concentration of H2SO4. Additionally, the photoelectrochemical properties of TiO2 depend on the nature of the electrolyte used. Other researchers reported that the photocatalytic activity of TiO2 can be greatly enhanced for films prepared by sulphuric acid treatment.22-24 Our experimental results agree well with the reported observation.
To find out about the surface morphology of the prepared TiO2 films, a scanning electron microscope (model: JSH-6700F FE-SEM) equipped with an EDAX X-ray micro analyser was used. The SEM micrographs of the samples prepared in 0.5 mol dm-3 H2SO4, by fixing the anodization time to 11 min and by varying the anodization voltage from 100 V to 150 V are shown in Fig. 3.
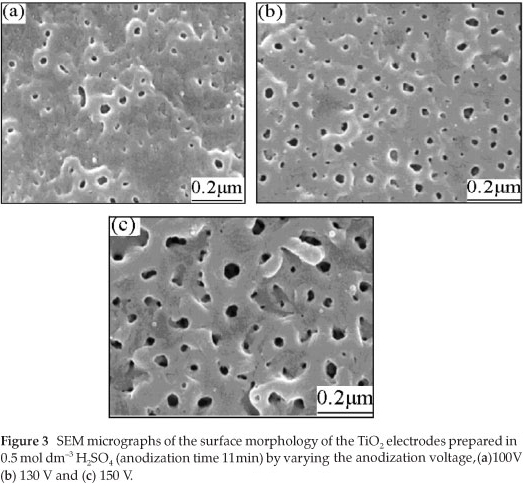
From Fig. 3, it can be observed that the film prepared at 100 V has more superficial pits with smaller pore density and pore diameter. The surface of the TiO2 film prepared at 130 V has a higher pore density and the pores are uniformly distributed. Although the film prepared at 150 V has a higher pore density, the pore diameter increased compared to the films prepared at 130 V and 100 V. The experimental result shows that the surface morphology largely depends on the anodization voltage, in agreement with the literature.25 From the surface morphology and the photoelectrochemical properties of the electrodes prepared in 0.5 mol dm-3 H2SO4 with an anodization time of 11 min, it was concluded that the anodization voltage of about 130 V could be the most suitable for preparing a TiO2 film with good photocatalytic properties. As a further piece of information, it is worth noticing that there were no pores formed on the surface of the film when anodizing was carried out at low voltages, i.e. at 60, 80, and 90 V (the corresponding micrographs are not shown here).
Figure 4 is related to the surface morphology of a TiO2 film prepared with NaOH as electrolyte.

From Fig. 4, it is observed that at low NaOH concentration and room temperature, the porous layer of the TiO2 film could not be produced on the titanium sheet. The films produced in this way are characterized by a rather rough surface and they are not multiporous compared to those prepared in H2SO4 electrolyte. This suggests that the electrolytic medium plays a significant role on the surface morphology of TiO2 prepared by anodic oxidation.
To find out about the crystallinity of the produced electrodes (A11,B11 and C11: refer to Table 1), the XRD of these samples was recorded. The XRD patterns of the prepared TiO2 electrodes after anodization at different experimental conditions are shown in Fig. 5.
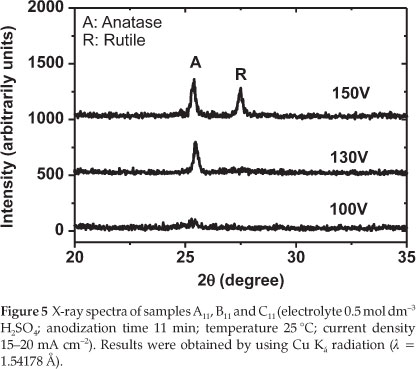
From Fig. 5, it can be seen that the film prepared at 100 V is mainly amorphous (neither anatase nor rutile could be detected). By increasing the anodization voltage to 130 V, the crystallization significantly increased. A strong peak related to anatase could be observed. Finally, when the oxidation voltage further increased to 150 V, a mixture of anatase and rutile could be observed. The experimental results demonstrated that the structure of the prepared TiO2 films responds sensitively to changes in voltage during anodizing of the titanium metal sheet. Therefore, in order to prepare TiO2 with anatase structure, it is necessary to fix the anodization voltage to 130 V. Figure 6 describes the XRD spectra of the electrodes J2,S2,S5 and S10 TiO2 thin films (see Table 2).
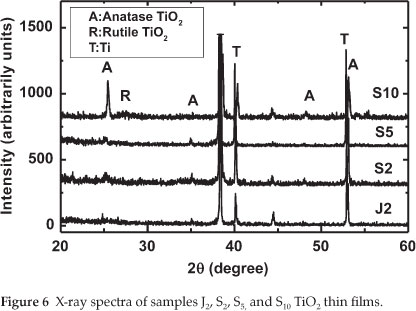
From Fig. 6, it can be seen that sample J2 prepared in NaOH (0.5 mol dm-3) was mainly amorphous. For the samples prepared in acid electrolyte (S2,S5 and S10), it is observed that, by increasing the H2SO4 concentration, the crystallization of TiO2 significantly increased and a strong peak related to anatase was observed (see sample S10 where H2SO4 concentration is 1.0 mol dm-3). This result indicates that the nature and the concentration of the electrolyte used in the production of TiO2 films plays an important role in its crystallization. The films prepared in NaOH display an amorphous structure, while those prepared in H2SO4 are crystalline.
From the surface morphology and the photoelectrochemical properties of the electrodes prepared in H2SO4 (0.5 mol dm-3) with an anodization time of 11 min, it was concluded that the anodization voltage of 130 V was the most appropriate for preparing electrodes with good photocatalytic properties.
Another experiment was run to investigate the influence of the H2SO4 concentration on the production of TiO2 thin films. For this purpose, the voltage was fixed at 130 V, the time set to 11 min and three concentrations of H2SO4 selected (0.5, 1.0 and 2.0 mol dm-3).
Figure 7 displays the XRD spectra related to the TiO2 films prepared under these different H2SO4 concentrations.
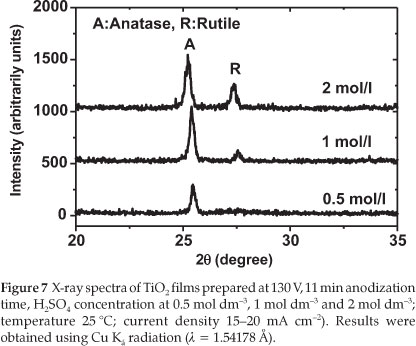
From Fig. 7, it can be seen that the TiO2 prepared in 0.5 mol dm-3 H2SO4 is crystalline and mainly in the anatase form whereas TiO2 prepared in 1.0 mol dm-3 H2SO4 consists mainly of anatase with some traces of rutile. When the concentration was increased to 2.0 mol dm-3, the TiO2 produced was a mixture of anatase and rutile. The amount of rutile increased with increasing H2SO4 concentration. The photocurrent of the prepared TiO2 film also decreased as shown in Table 3. This result indicates that the photocatalytic activity depends significantly on the crystallinity of the TiO2 film prepared, and on the amount of rutile present in the sample. The sample prepared with 1.0 mol dm-3 H2SO4 exhibited a higher value of the photocurrent density, in good agreement with the result reported by other researchers who observed that a mixture of anatase and a small amount of rutile leads to a higher catalytic activity compared to a single crystal of anatase.26-28
Finally, the effect of the anodization time on the preparation of TiO2 thin films was investigated. For this purpose, the oxidation voltage was fixed to 130 V, the concentration of H2SO4 to 1.0 mol dm-3, and three anodization times selected (5, 8 and 11 min) as shown in Tables 2 and 3.
From Table 3, it can be seen that the photocurrent obtained for sample E5 was the highest with a value IUV = 6.5 mA cm-2. An increase in the anodization time showed a significant decrease in the photocurrent densities (refer to samples E8 and E11 in Table 1). Moreover, the crystallinity of sample E11 (prepared in 1.0 mol dm-3 H2SO4,130 V and 11 min) was mainly constituted of anatase TiO2 with small traces of rutile. The particle size calcu- lated from the XRD spectrum by using the Scherrer formula was about 23 nm.29-30 Figure 8 shows the XRD spectrum of the TiO2 prepared under the same conditions except that the oxidation time was fixed to 5 min (sample E5).
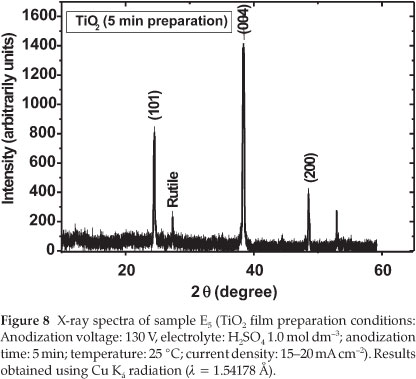
The size of TiO2 prepared in 5 min was evaluated by using the Scherrer formula, and the average size calculated from the peak (101) at 2θ of 25.28 °C related to anatase is about 14.6 nm. This means that the TiO2 film produced in 1.0 mol dm-3 H2SO4,5 min after the anodization at 130 V has the smallest diameter compared to those prepared at 8 and 11 min. This result indicates that the anodization time fixed in the production of TiO2plays an important role in its photocatalytic activity.
4. Conclusion
From the above results, it appears that, in order to prepare multinanoporous TiO2 films with good photocatalytic properties, the best experimental conditions for anodization would be as follows: 130 V for the anodization voltage, 5 min for the anodization time, and 1.0 mol dm-3 for the H2SO4 electrolyte concentration.
Compared with conventional techniques like plasma spray or sol-gel, the present work shows that the nanostructure of the TiO2 films (with diameter size less than 100 nm) can be easily obtained at lower cost by electrochemical oxidation on a titanium metal substrate. Furthermore, the structures and morphologies of the TiO2 films can be controlled according to the anodizing conditions. The easy control of microstructures of the anodic film should be a major advantage for many technical applications especially in wastewater treatment.
Acknowledgements
The authors gratefully thank Dr. Xin Lihui of the National Center of Shanghai Institute of Measurement and Testing Technology for his help with SEM, TEM, Raman and X-ray analysis.
References
1 K. Wessels, A. Feldhoff, M. Wark, J. Rathousky and T. Oekermann, Electrochem. Solid-State Lett, 2006, 9, 93-96. [ Links ]
2 S. Teekateerawej, J. Nishino and Y. Nosaka, J. Photochem. Photobiol. A, 2006, 179, 263-268. [ Links ]
3 D. Fabbri, L.S. Villata, A.B.Prevot, A.L. Capparelli and E. Pramauro, J. Photochem. Photobiol .A, 2006, 180, 157-164. [ Links ]
4 C.C. Chen, C.S. Lu and Y.C. Chung, J. Photochem. Photobiol. A, 2006, 181, 120-125. [ Links ]
5 C.G. da Silva, W. Wang and J.L.Faria, J. Photochem. Photobiol. A, 2006, 181, 314-324. [ Links ]
6 S. Somasundaram, Y. Ming, C.R. Chenthamarakshan, Z.A. Schelly and K. Rajeshwar, J. Phys. Chem. B, 2004, 108, 4784-4788. [ Links ]
7 A. Burns, W. Li, C. BakerandS.I. Shah, Proc. Mater. Res. Soc. Sym.,2002, 703, 193-197. [ Links ]
8 C.A. Morris, M. L. Anderson, R.M. Stroud, C.I. Merzbacher and D.R. Rolison, Science, 1999, 284, 622-624. [ Links ]
9 L. Davydov, F.P. Amaat and S.P. George, Photocatalytic Degradation of organic Compounds, U.S Patent No. 6585863, 1 July 2003. [ Links ]
10 D. Byun, Y. Yin, B. Kim, J. Lee and D. Park, J. Hazard. Mater, 2000, 73, 199-206. [ Links ]
11 H. Noguchi, R. Kagami, M. Kuzumi and S. Shigeo, Inventors, Apparatus for Continual Passing Water for Treatment using Ozone and Photocatalyst, Meidensha Electric Mfg, Co., Ltd, Japan, assignee, Japan Patent, 2001170498.20010626.135:68462 CA, 2001. [ Links ]
12 O. Chihiro, A. Shigendo, O.Yoshiaki, H. Tatsuro, K. Susuki, K. Shin-Ichi, H. Yoshida and H. Tadashi, J. Mater. Chem., 1999, 9, 2943-2952. [ Links ]
13 M. Antonietti, R.A. Caruso, H.P. Hentze and C. Göltner, Hydrophilic gels with new superstructures and their hybrids by nanocasting technologies, Macromol. Symp. (Polymers Friendly for Environment), 2000, 152, 163-172. [ Links ]
14 G. Qing-Ping W. Zhang-Huang, Z. Jun-Fu and T. Fang, J. Nat. Gas Chem., 1999, 8, 331-338. [ Links ]
15 K. Kiyoshi, Y. Murata, I. Tawara and K.Takeuchi, Air purification pavement: Development of photocatalytic concrete blocks, Proc. Beijing Int. Symp. Cem. Concr., International Academic Publishers, 4th edn., Beijing, Peop. Rep. China, 1998, 751-755. [ Links ]
16 Y. Matsumoto, M. Murakami, Z.W. Jin, A. Nakayama, T. Yamagushi, T. Ohmori, E. Susuki, S. Nomura, M. Kawasaki and H. Koinuma, Combinatorial synthesis and high through put evaluation of doped TiO2 thin films for the development of photocatalysts, in Combinatorial and Composition Spread Techniques in Materials and Device Development, Proc. SPIE-Int. Soc. Opt. Eng. 3941, 2000, 19-27. [ Links ]
17 D. Makoto, S. Saka, H. Miyafuji and A.I. David, Mater. Sci. Res. Int., 2000, 6, 15-21. [ Links ]
18 C. Massaro, P. Rotolo, F. De Riccardis, E. Milella, A. Napoli, , M. Wielarld, T.M. Spencer and D.M. Brunette, J. Mater. Sci. Mater. Med, 2002, 13, 535-548. [ Links ]
19 R. Palombari, M. Ranchella, C. Rol and G.V Sebastiani, Sol. Energ. Mat. Sol. Cell., 2002, 71, 359-368. [ Links ]
20 G.A. Ragoisha and A.S. Bondarenko, Electrochemistry: New Research Potentiodynamic Electrochemical Impedance Spectroscopy, (M. Nunez, ed.), Nova Science Publ., New York, 2005, pp. 51-75. [ Links ]
21 J.C. Myland and K.B. Oldham, Anal. Chem., 2000, 72, 3210-3217. [ Links ]
22 J.C. Yu, J.G. Yu and J.C. Zhao, Appl. Catal. B, 2002, 36, 31-43. [ Links ]
23 H. Okuyama, K. Honma and S. Ohno, J. Japan Inst. Metals, 1999, 63, 74-81. [ Links ]
24 L. Zhao, Y. Yu, L. Song, X. Hu and A. Larbot, Appl. Surf. Sci., 2005, 239, 285-291. [ Links ]
25 V.M. Frauchiger, F. Schlottig, B. Gasser and M. Textor, Biomaterials, 2004, 25, 593-606. [ Links ]
26 T.E. Doll and F.H. Frimmel, Water Res., 2005, 39, 847-854. [ Links ]
27 H. Ma, L.F. Liu, F.L. Yang, X.W. Zhang and J.M. Yu, Photogr. Sci. Photochem., 2006, 24, 17-27. [ Links ]
28 J. B. Wachtman and H. K. Zwi, Characterization ofMaterials, Manning Publications Co., Butterworth-Heinemann, Greenwich, USA, 1993. [ Links ]
29 B.D. Cullity, Elements of X-ray Diffraction, Addison-Wesley Publishing Co., London, England, 1959. [ Links ]
Received 13 December 2011
Revised 14 November 2012
Accepted 28 November 2012
* To whom correspondence should be addressed. E-mail: jeremiemuswem@yahoo.fr


{kind=link}