










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Animal Science
On-line version ISSN 2221-4062
Print version ISSN 0375-1589
S. Afr. j. anim. sci. vol.52 n.2 Pretoria 2022
http://dx.doi.org/10.4314/sajas.v52i2.9
Diversity and taxonomic identification of bacteria in the rumen of zebu cattle fed various diets
B.K. KorirI, #; J.K.N. KuriaII; D.M. MwangiI; C.K. GachuiriII
IKenya Agricultural and Livestock Research Organization, P. O. Box 12-90138, Makindu, Kenya
IIFaculty of Veterinary Medicine, University of Nairobi
ABSTRACT
In this study bacterial populations were identified in the rumen of zebu cattle fed various diets and classified taxonomically with metagenomic sequencing of the 16S rRNA gene. Twenty-four (24) heifers were used in a completely randomized experimental design to test the effect of the diets. Treatment 1 consisted of range grass hay. Treatment 2 was composed of the hay diet augmented with sun-dried cassava leaves. Treatment 3 comprised hay plus sun-dried azolla. Treatments 4 to 6 were similar to treatments 1 to 3. but with a basal diet of Brachiaria Mulato II hay. Rumen liquor samples were collected from the heifers, from which a total of 192 DNA samples were amplified and the resulting 16S rRNA sequences were compared with those in the National Centre for Biotechnology Information BLAST database using MetagenAssist. Bioinformatics analyses indicated that 17 operational taxonomic units (OTUs) were present at phylum level, of which 43.3% were Firmicutes, 27.2% Bacteroidetes, 22.8% Proteobacteria and 1.7% Euryarchaeota. The remaining OTUs were Cyanobacteria (1.4%) and Chloroflexi (1%) with Actinobacteria, Verrucomicrobia, Spirochaetes, Tenericutes, Planctomycetes, Elusimicrobia, Lentisphaerae, Armatimonadetes, Fibrobacteres, Synergistetes and Arthropoda all below 1% of the organisms present. Time and diet both affected (P<0.05) the abundance of microbes, but not their diversity in the rumen. Thus, these diets could affect the performance of animals.
Keywords: 16S rRNA, communal composition, diets, microbial diversity, ruminal contents
Introduction
There is increasing demand for cattle products as a result of continued population growth and improving standards of living in developing countries. There is therefore a need to improve feed utilization. Rumen microbiome studies could inform identification of new ways to ensure efficient use of feed and improved animal health. The microorganisms in the rumen form a complex ecosystem that is well suited to diverse diets (Clauss et al., 2010, Stevens & Hume, 1998). A symbiotic relationship has developed over time between ruminants and these ruminal microorganisms, which enables the digestion of fibrous materials (Dehority & Orpin, 1997). The relationships between microorganisms and the host animal affect their performance (Rawls et al., 2004). In the rumen, the microorganisms receive substrates through ingested feed. Through fermentation, these microorganisms become a valuable source of nutrients for the host animal (Mizrahi, 2013).
Techniques based on 16S rRNA made it possible to analyse complex ecosystems, and to determine the diversity and abundance of communities of microorganisms (Amann et al., 1995). Metagenomics made it possible to identify uncultured microorganisms, their evolution, and functional relationships (Thomas et al., 2012, Sabree et al., 2009). The 16S rRNA gene, an excellent phylogenetic marker (Pace, 1997) consists of regions that are highly variable and conserved, and their differences in sequences differentiate the microorganisms and determine their phylogenetic relationships. rRNA gene fragments can be obtained without cultivating the microorganisms from 16S rDNA libraries. This is achieved by amplifying 16S rRNA, obtained from samples, with polymerase chain reaction (PCR). In this way, a list of 16S rRNA genes is developed. The composition of the microbes can be determined through sequence analysis and comparing the analysis with appropriate reference sequences in databases to infer their phylogenetic affiliation (Illumina, 2013).
Feeding fibrous materials to ruminants can affect ruminal microbial communities and increase ammonia nitrogen (Saro et al., 2012). Ruminants convert light energy captured by plants into edible compounds such as milk and meat (Nathani et al., 2015). The objective of this study was to identify and document the composition of rumen bacteria in zebu heifers fed various types of diet through metagenomics sequencing of the 16S rRNA gene.
Materials and Methods
The feeding trial was carried out at Kenya Agricultural and Livestock Research Organization (KALRO) Kiboko Research Centre in Makindu sub-County, Makueni County. The laboratory work was done at the International Livestock Research Institute (ILRI), Nairobi. The procedures were approved by the committee in charge of animal care and use at KALRO. A completely randomized design experiment was used in the trial, which ran for 14 weeks, during which twenty-four (24) yearling small East African short-horned zebu heifers were used with mean live weight of 109.8 ± 18.4 kg. The heifers were randomly assigned to 12 pens with each pen housing two heifers. Six dietary treatments were then allocated to two pens each, so that four heifers were allotted to each treatment.
Treatment 1 consisted of a mixture of range grass hays (mainly Eragrostis superba). Treatment 2 consisted the hay plus sun-dried cassava leaves. Treatment 3 comprised the hay plus sun-dried azolla. Treatments 4 to 6 were similar to treatments 1 to 3, but with Brachiaria Mulato II hay as the basal diet. The crude protein content of the basal feeds was used to determine the amount of supplement for each animals to provide 16% CP, as recommended for growing animals (NRC, 2000).
The animals were first offered the supplementary azolla or cassava leaves at 08h00. In general, the animals preferred the grass hay to the supplementary materials. Half of the amount of hay allocated to the animal was given at 10h00 and the other half at 14h00 hours to minimize wastage. Intake was determined by collecting and weighing the leftovers before the next day's feeding. The feed was placed in two plastic basins in each enclosure and water was provided ad libitum in two buckets. A mineral block (Afya Bora® stock lick) was provided ad libitum. Ticks and flies were controlled biweekly with a pour-on acaricide (Ectopor 020 SA). All the animals were treated with 10% Albendazole suspension before the start of the experiment to control internal parasites.
Samples of the feeds were put in bags and dried in the oven for 48 hours at 60 °C to determine air-dry matter. The samples were ground through a 1.0 mm sieve in a hammermill and retained for nutritional analysis. They were then dried overnight in the oven at 105 °C to determine their dry matter content. Crude protein was determined as the Kjeldahl nitrogen content multiplied by 6.25 (AOAC, 2005). Procedures described in Van Soest et al. (1991) were used to determine acid detergent lignin and neutral detergent fibre. The samples were burned in a muffle furnace at 550 °C for 8 hours to determine the ash content.
Rumen fluid was collected from all the animals on day 14, first in the morning before feeding, and then every other hour for seven hours. The animals were restrained in a chute and a flexible stomach tube was inserted through the mouth. An attached suction pump was used to withdraw the samples, which were put in 10 mL cryotubes and placed in liquid nitrogen until the DNA was extracted.
Extraction of DNA was done with the Zymo soil extraction kit (ZYMO Research, California, USA) following the protocol of the manufacturer. Briefly, 150 uL of rumen liquor was added to a ZR BashingBead™ lysis tube followed by 750 μL of solution for lysis. The lysis tube was then fixed in a bead beater, which had tube holder assembly (Disruptor Genie™), followed by processing for 5 min at maximum speed. The same lysis tube was centrifuged for 1 min at 10 000 x g. Then 400 μL of the supernatant was placed in a Zymo-Spin™ IV spin filter and centrifuged for 1 min at 7000 x g. Then 1,200 μL of buffer for binding was ten mixed with the flow through. Then 800 μL of the mixture was placed in a Zymo-Spin IIC column and centrifuged for 1 min at 10 000 x g. This step was repeated after discarding the flowthrough. When extraction had been completed, DNA quality was confirmed by electrophoresis on a 0.8% agarose gel visualized under ultraviolet light.
The primers used for PCR amplification and those for sequencing were similar to those used by Caporaso et al. (2010a). This supported pooling of up to 2167 samples in each lane. Both PCR primers (515F and 806R) contained sequences of the Illumina flow cell adapter regions. The V4 region of 16S rRNA was amplified with 515F universal primers (5'-AGAGTTTGATCMTGGCTCAG -3'), and 806R (5'-CGGTTACCTTGTTACGACTT-3') (Caporaso et al , 2010b). The amplification mixture consisted of 10 x Dream Taq buffer (2.5 μL, 10 mM dNTPs (0.5 μL), 10 nM of each primer, DNA template (1 μL) and 0.2 μL of DreamTaq polymerase (Thermo Fisher Scientific, Massachusetts, USA). The amplification was carried out on a thermo cycler (Applied Biosystems, Fisher Scientific, Massachusetts, USA) using the manufacturer's protocol. Amplified fragments were visualized on a 0.8% agarose gel under ultraviolet light (Figure 1). The PCR amplicons were purified on a QIAquick DNA gel extraction kit (QIAGEN, CA).
The concentration of samples for sequencing was determined with Qubit® dsDNA BR assay kit (Thermo Fisher) before sequencing with the Illumina platform. Filtering of reads for quality was done and truncated at their first low-quality base. Any reads that had 75 bases and below were dismissed, similar to those that had barcodes that did not match those expected. Apart from the 192 experimental samples, the MiSeq run, as part of the control, had phiX174, which accounted for 47% of the reads in this run. The limited diversity among the 16S amplicons was the reason for the use of the phiX control.
The assigning of reads to OTUs was done with a closed-reference protocol, as described in the QIIME toolkit (Caporaso et al., 2010a), in which UCLUST algorithm (Edgar, 2010) was used in the search and comparison of sequences with the Greengenes database (Desantis et al., 2006), filtering them at an identity of 97%. The best hit approach was used to assign reads to the OTUs. Reads that were not similar to any reference sequence were dismissed. A dominant OTU was defined as consisting of < 1% of total sequences, and only dominant OTUs were included in the analysis. A dominant or non-dominant OTU was considered prevalent across a group if it had sequences from all samples in that group. It was also assumed that prevalent and dominant OTU or groups could be considered part of the bacterial core.
SPSS (v 20) (9IBM Corp., Armonk, NY, USA) software was used for statistical analyses. Shannon indices were compared using the Student's t-test.
Results and Discussion
Table 1 shows the results of sequencing amplicons using the Illumina platform. In total 19,430,463 reads were generated before quality control. This was reduced to 11 838,743 after duplicates were removed. When filtered by length and error, 3 149 693 reads remained and these gave rise to 2,929 OTUs.
Bacterial community composition indicated seventeen bacterial phyla, with three, namely Firmicutes (43.3%), Bacteroidetes (27.2%), and Proteobacteria (22.9%), being the most abundant (Figure 2). The other 14 phyla each represented <2% of all the bacterial sequences, namely Euryarchaeta, Chíorofíexi, Cyanobacteria, Actinobacteria, Verrucomicrobia, Spirochaetes, Tenericutes, Planctomycetes, Elusimicrobia, Lentisphaerae, Armatimonadetes, Fibrobacteres, Synergistetes and Arthropoda. The abundance of Firmicutes in the six treatments was 49.8%, 44.3%, 39.1%, 42.0%, 44.7% and 41.5% (SE = 0.02%), respectively. Proteobacteria was most abundant in heifers treated with cassava leaf meal (26.5%), followed closely by heifers treated with azolla at 26%. For the other diets, the abundance of Proteobacteria was 23.9% and 19.2% for Bracharia, and the control diet. The abundance of Bacteroidetes phylum was highest in the heifers treated with Bracharia and cassava leaf meal at 30%. This was followed by Bracharia alone, cassava leaf meal alone, and the control diet at 28.5%, 26.9% and 25.9% respectively.
The abundance of some phyla was affected significantly by the feed, whereas others were not affected (Table 2). Proteobacteria, Actinobacteria, Spirochaetes, Bacteroidetes, Euryarchaeta and Armatimonadetes were affected significantly by feeds. Bracharia had the most significant effect on the abundance of various phyla, followed by azolla and cassava leaf meal.
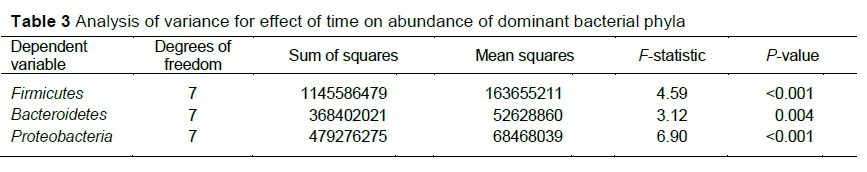
The abundance of Firmicutes, Proteobacteria and Bacteroidetes (P <0.05) was affected by time (Table 3). The change in the structure of the rumen microbes over time was interesting. It indicated that if animals were given time, they adapted to newly introduced feeds and thus could improve feed utilization efficiency.
Shown in Figure 3 are time trends in the Shannon diversity index as affected by the diets. Diversity of species in a community structure expresses the characteristics that are peculiar to that community level of organization. A higher value of the Shannon index of diversity indicates a more diverse community of species, whereas a lower value indicates a less diverse community. An index value of 0 indicates a monoculture. The Shannon index of diversity in this study (2.6) indicated that the diversity of bacterial species was high. Thus, these feedstuffs enabled the coexistence of a range of rumen microbes and suggested that these diets would not alter the community of microorganisms in the rumen of these heifers.
Bacteroidetes and Firmicutes were most common phyla. This was similar to work by Edwards et al. (2004), which showed them to be dominant in the rumen. There were other large groups of bacteria that were not classified, including Clostridiales, Lachnospiraceae, and, Ruminococcaceae, which were likely to be present in the rumen. However, members of Bacteroidetes and Firmicutes are frequently the most abundant bacteria detected in the rumen by culture-independent methods. In this study, the prevalence of Firmicutes was 82.1%, which is one of the highest values reported for the rumen. Other studies recorded values of 90.2% and 95% of sequences assigned to Firmicutes in Holstein cows on high roughage and grain diets (Tajima et al., 2000). However, most studies reported values that were lower than 70%.
In addition, Cyanobacteria was detected in the samples, irrespective of the diet. This phylum is known for its photosynthetic capability, but recent studies demonstrated the presence of non-photosynthetic members in the stomach of humans and in underground water (Di Rienzi et al., 2013). Their analysis demonstrated that the order YS2, present in the current data, had roles such as obligate anaerobic fermentation, fixation of nitrogen, production of hydrogen syntrophically and the manufacture of vitamins K and B. YS2 has been suggested as a new phylum 'Melainabacteria' (Di Rienzi et al., 2013). Other reports showed its presence in the gut of mammals (Soo et al., 2014; Zeng et al., 2015). Currently work that described the microbiome of zebu heifers is limited and more high-throughput sequencing studies are necessary. Pitta et al. (2016), showed the abundance of Bacteroidetes, Firmicutes and Proteobacteria szx was 70%, 15-20% and 7% respectively. This dominance was influenced by diet and the age of the cow. Additionally, studies by Alzahal et al. (2017) demonstrated structural and composition differences in the bacterial microbiome between dry and cows in early lactation. The lactating cows had greater proportions of Proteobacteria, lower amounts Firmicutes, and no change in the proportion of Bacteroidetes.
Conclusions
Rumen microbes in zebu cattle are highly diverse and their abundance is affected by their feedstuffs. Time after feeding affected the structure of the bacterial community, but not microbial diversity. The results showed that it is possible to introduce feeds such as azolla to ruminants and they would be able to utilize them.
Acknowledgement
The authors acknowledge the European Union Arid and Semi-Arid Lands Agricultural Productivity Research Project (ASAL APRP) for funding the study. This work was carried out at KALRO Kiboko and the support of the institute director, the centre director and entire staff of Kiboko is appreciated. The Biosciences Eastern and Central Africa International Livestock Research Institute, BecA-ILRI Hub co-funded the fellowship to carry out DNA extraction and sequencing work.
Authors' Contributions
BKK was responsible for running the trial, performing laboratory analysis and data processing and interpretation. BKK wrote the manuscript, which was reviewed by JKNK, CKG and dMm.
Conflict of Interest Declaration
There are no conflicts of interest.
References
Alzahal, O., Li, F., Guan, L.L., Walker, N.D. & McBride, B.W., 2017. Factors influencing ruminal bacterial community diversity and composition and microbial fibrolytic enzyme abundance in lactating dairy cows with a focus on the role of active dry yeast. J. Dairy Sci. 100, 4377-4393. https://doi.org/10.3168/jds.2016-11473 [ Links ]
Amann, R., Ludwig, W. & Scheifer, K.H., 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59(1), 143-69. [ Links ]
AOAC, 2005. Official methods of analysis, 18th edition. AOAC International, Rockville, Maryland. USA. [ Links ]
Caporaso, J.G., Lauber, C.L., Walters, W.A., Berg-Lyons, D., Lozupone, C.A., Turnbaugh, P.J., Fierer, N. & Knight R., 2010b. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS 108 (supplement 1), 4516-4522. https://doi.org/10.1073/pnas.1000080107 [ Links ]
Caporaso, J.G, Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., Fierer, N., Pena, A.G., Goodrich, J.K., Gordon, J.I., Huttley, G.A., Kelley, S.T., Knights, D., Koenig, J.E., Ley, R.E., Lozupone, C.A., McDonald, D., Muegge, B.D., Pirrung, M., Reeder, J., Sevinsky, J.R., Turnbaugh, P.J., Walters, W.A., Widmann, J., Yatsunenko, T., Zaneveld, J. & Knight, R., 2010a. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335-336. DOI: 10.1038/nmeth.f.303 [ Links ]
Clauss, M., Hume, I.D. & Hummel, J., 2010. Evolutionary adaptations of ruminants and their potential relevance for modern production systems. Animal 4(7), 979-92. DOI: 10.1017/S1751731110000388 [ Links ]
Dehority, B.A. & Orpin, C.G., 1997. Development of and natural fluctuations in rumen microbial populations. In P.N. Hobson & C.S. Stewart (eds). The rumen microbial ecosystem., 2nd edition. Blackie Academic & Professional, London. Pp. 196 245. [ Links ]
DeSantis, T.Z., Hugenholt, P., Larsen, N., Rojas, M., Brodie, E.L., Keller, K., Huber, T., Dalevi, D., Hu, P. & Andersen, G.L., 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72(7), 5069-72. DOI: 10.1128/AEM.03006-05 [ Links ]
Di Rienzi, S.C., Sharon, I., Wrighton, K.C., Koren, O., Hug, L.A., Thomas, B.C., Goodrich, J.K., Bell, J.T., Spector, T.D., Banfield, J.F. & Ley. R.E., 2013. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. eLife 2, e01102. DOI: 10.7554/eLife.01102 [ Links ]
Edgar, R.C., 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19), 2460-2461. DOI: 10.1093/bioinformatics/btq461 [ Links ]
Edwards, J.E., McEwan, N.R., Travis, A.J., & Wallace, R.J., 2004. 16S renal library-based analysis of ruminal bacterial diversity. Anton. Leeuw. Int. J. G. 86, 263-281. [ Links ]
Illumina, 2013. 16S metagenomics sequencing library preparation. https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf [ Links ]
Mizrahi I., 2013. Rumen symbioses. In: Rosenberg E., DeLong E.F., Lory S., Stackebrandt E. & Thompson F. (editors) The Prokaryotes. Springer, Berlin, Heidelberg, Germany. https://doi.org/10.1007/978-3-642-30194-0_1 [ Links ]
Nathanil, N.M., Patel, A., Mootapally, C.S., Reddy, B., Shah, S.V., Lunagaria, P.M., Kothari, R.K. & Joshi, C.G., 2015. Effect of roughage on rumen microbiota composition in the efficient feed converter and sturdy Indian Jaffrabadi buffalo (Bubalus bubalis). BMC Genomics (2015) 16:1116. DOI: 10.1186/s12864-015-2340-4 [ Links ]
NRC (National Research Council), 2000. Nutrient requirements of beef cattle. 7th revised edition. NRC, Washington DC, USA. [ Links ]
Pace, N.R., 1997. A molecular view of microbial diversity and the biosphere. Science 276, 734-740. DOI: 10.1126/science.276.5313.734 [ Links ]
Pitta, D.W., Indugu, N., Kumar, S., Vecchiarelli, B., Sinha, R., Baker, L.D., Bhukya, B. & Ferguson. J.D., 2016. Metagenomic assessment of the functional potential of the rumen microbiome in Holstein dairy cows. Anaerobe 38:50-60. DOI: 10.1016/j.anaerobe.2015.12.003 [ Links ]
Rawls J.F., Samuel B.S. & Gordon J.I., 2004. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. PNAS 101 (13), 4596-4601. DOI: 10.1073/pnas.0400706101 [ Links ]
Rosenberg, E., DeLong, E.F., Lory, S. Stackebrandt, S. & Thompson, F. (eds). The prokaryotes. Prokaryotic biology and symbiotic association. SpringerLink. DOI: 10.1007/978-3-642-30194-0 [ Links ]
Sabree, Z.L., Rondon, M.R. & Handelsman, J., 2009. Metagenomics. In: Schaechter, M. (editor), Encyclopedia of Microbiology (2nd ed.). pages 622-632). Academic Press, Cambridge, Massachusetts, USA. https://handelsmanlab.discovery.wisc.edu/wp-content/uploads/2018/01/Sabree-Rondon-Handelsman-Metagenomics.pdf [ Links ]
Saro, C., Ranilla, M. J. & Carro, M.D., 2012. Postprandial changes of fiber-degrading microbes in the rumen of sheep fed diets varying in type of forage as monitored by real-time PCR and automated ribosomal intergenic spacer analysis. J. Anim. Sci. 2012, 90, 4487-4494. DOI: 10.2527/jas.2012-5265 [ Links ]
Soo, R.M., Skennerton, C.T., Sekiguchi, Y., Imelfort, M., Paech, S.J., Dennis, P.G., Steen, J.A., Parks, D.H., Tyson, G.W. & Hugenholtz, P., 2014. An expanded genomic representation of the phylum cyanobacteria. Genome Biol. Evol. 6,1031-1045. DOI: 10.1093/gbe/evu073 [ Links ]
Stevens, C.E. & Hume, I.D., 1998. Contributions of microbes in vertebrate gastrointestinal tract to production and conservation of nutrients. Physiol. Rev. 78(2, 393-427. DOI: 10.1152/physrev.1998.78.2.393 [ Links ]
Tajima, K., Arai, S., Ogata, K., Nagamine, T., Matsui, H. & Nakamura, M., 2000. Rumen bacterial community transition during adaptation to high-grain diet. Anaerobe 6(5, 273±84. https://doi.org/10.1006/anae.2000.0353 [ Links ]
Thomas, T., Gilbert, J. & Meyer, F., 2012. Metagenomics - a guide from sampling to data analysis. Microb. Inform. Exp. 2(3). http://dx.doi.org/10.1186/2042-5783-2-3. [ Links ]
Van Soest, P.J. Robertson, J.B. & Lewis B.A., 1991: Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci. 74, 3583-3597. DOI: 10.3168/jds.S0022-0302(91)78551-2 [ Links ]
Zeng, B., Han, S., Wang, P., Wen, B., Jian, W., Guo, W., Yu, Z., Du, D., Fu, X., Kong, F., Yang, M., Si, X., Zhao, J. & Li. Y., 2015. The bacterial communities associated with fecal types and body weight of rex rabbits. Sci. Rep. 5, 9342. DOI: 10.1038/srep09342 [ Links ]
Submitted 19 January 2020
Accepted 17 January 2022
Published 27 April 2022
# Corresponding author bernardkorir@gmail.com


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}