










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Animal Science
versão On-line ISSN 2221-4062
versão impressa ISSN 0375-1589
S. Afr. j. anim. sci. vol.52 no.2 Pretoria 2022
http://dx.doi.org/10.4314/sajas.v52i2.4
ARTICLES
Rumen microbial diversity of Bonsmara cattle using amplicon sequencing during a 120-day growth trial
D.A. LindeI, #; E. van Marle-KösterI; C.J.L. du ToitI; M.M. ScholtzII; D. SchokkerIII
IDepartment of Animal Science, University of Pretoria, Pretoria, South Africa, 0043
IIAgricultural Research Council Animal Production, Agricultural Research Council, Irene, South Africa, 0062
IIIWageningen Livestock Research, Wageningen University and Research, Wageningen, The Netherlands, 3386700
ABSTRACT
Improved understanding of the microbial populations during intensive feeding of feedlot cattle holds potential for optimizing production efficiency. Ionophores are used to increase the production and efficiency of ruminants and are commonly used in South African feedlots. Bonsmara bull calves (n=24) were subject to a four-phase feedlot diet in a growth trial commencing with backgrounding, followed by starter, grower and finisher diets. Animals were randomly divided into two groups: control and a group provided the in-feed ionophore monensin. Four animals from each group were randomly selected for rumen content collection using an oesophageal tube during the phases in the trial. Samples were analysed using 16S rRNA and internal transcribed spacers amplicon sequencing. Totals of 42 008 and 35 442 amplicon sequence variants were identified from 16S rRNA and internal transcribed spacers amplicon sequencing. The rumen microbiome composition and alpha diversity differed significantly between the phases, whereas no significant difference was observed between the control and monensin groups. Backgrounding had the highest bacterial richness, whereas the grower phase had the highest fungal richness. Bacteroidetes, Firmicutes and Proteobacteria were the most abundant phyla, with Bacteroidetes being most abundant in the backgrounding and starter phases, whereas Proteobacteria was the most abundant in the grower and finisher phases. Ascomycota, Basidiomycota and Neocallistigomycota were the most abundant fungal phyla. Improved knowledge of the shift in microbiome population during the growth period could assist in adapting feeding strategies to improve the efficiency of beef production.
Key words: feedlot, microbial shift, rumen microbiome, ruminant
Introduction
Beef producers are presented with the challenge of increasing the supply of high-quality beef, while maintaining an economic and environmentally sustainable enterprise. A potential solution is to increase the feed efficiency (Capper, 2011). South African beef production is characterized by medium to large extensive commercial cow-calf operations with 65% to 70% of all cattle that are slaughtered having originated from feedlot systems (DAFF, 2019). Several factors play a role in economic beef production, such as the feeding regime, price of weaners, and general health and management of the animals. Overall, feed efficiency is the determining factor for sustainable feedlot production (Koenig et al., 2020).
The feed efficiency is determined by various factors, including the rumen microbiome (Guan et al., 2008; Myer et al., 2015). Microbes (Firkins & Yu, 2015) are responsible for fermentation and degradation of the feed components into nutrients such as volatile fatty acids (VFAs) that provide approximately 70% of the energy available to the animal for its maintenance and production (Perea et al., 2017). The composition and balance of the rumen microbiome determines the concentration of the VFAs and thus greatly influences the energy that is available for the animal to metabolise.
In South Africa, the efficiency and production of feedlot animals are increased by adding ionophores, such as monensin, to the diet and thereby changing the rumen microbiome by inhibiting gram-positive bacteria and methanogens and shifting in VFA production in favour of propionate (Samuelson et al., 2016). Propionate is glucogenic, thus providing more energy to the animal and thus increasing its efficiency.
Transitioning from a roughage-based diet to a concentrate-based diet has been reported to modify the rumen microbiome (Fernando et al., 2010; Stanton et al., 2020), with a sudden transition resulting in digestive disorders (Klieve et al., 2003). A stepwise adaptation from a roughage diet to a high energy one is known to stabilize the rumen microbiome (Klieve et al., 2003; Bevans et al., 2005). These feeding regimes are characteristic of feedlot feeding and therefore necessitate improved understanding of the dynamics of the rumen microbiome (Mackie et al., 1978; Tajima et al., 2001). Improving feed efficiency in the livestock sector is crucial to sustainable animal production as it could improve nutrient utilization from feed, increase profitability, and reduce greenhouse gas emissions (Huws et al., 2018).
Modern sequencing-based methods, such as 16S rRNA and internal transcribed spacer (ITS) amplicon sequencing, have improved the detection and quantification of microbes in the rumen. Previous methods such as culturing could not capture the full diversity of the rumen microbiome (Huws et al., 2018; Gruninger et al., 2019). These sequencing techniques can be used to conduct research on the total microbial diversity and microbial population function (Myer, 2019) and could lead to improved understanding of the interaction between the diet, the rumen microbiome, and efficiency of production (Pitta et al., 2018).
In this study, it was hypothesized that the bacterial, archaeal and fungal populations in the rumen of Bonsmara cattle will differ across the phases in the feedlot period and in response to the feeding of monensin.
Materials and Methods
The Animal Ethics Committee of the University of Pretoria granted approval for the project (NAS445/2019). The trial was conducted on Sernick Group (Pty) LTD Farm, Edenvale, Free State, South Africa. Twenty-four Bonsmara bull calves (10-12 months old, 228 ± 22 kg) from a single Bonsmara breeder were backgrounded for 40 days on veldt grazing with lick supplementation before the start of the growth period. The animals were divided into two treatment groups: a group receiving a standard feedlot diet that included 30 mg/animal/day of monensin (n=12); and a control group that was fed the same diet without monensin (n=12). The diets were mixed at Sernick feed mill, marked and bagged for the trial.
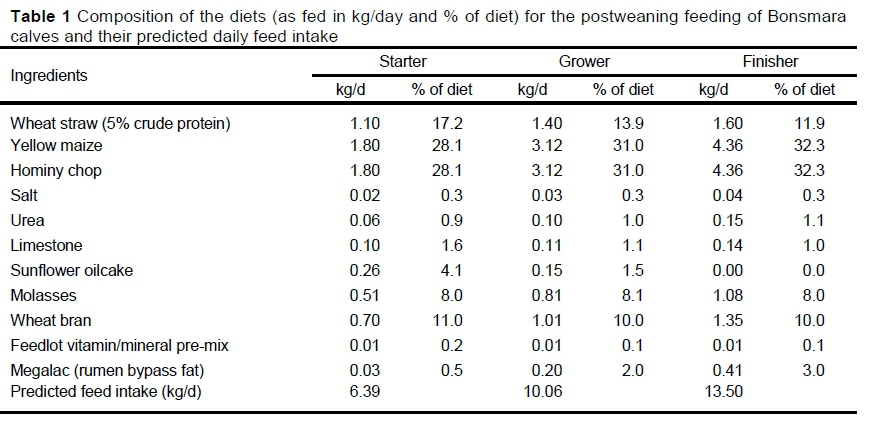
The animals were allocated in a randomized block design according to weight, three to a pen, with eight pens in total for the growth study, which consisted of starter, grower, and finisher phases. The composition of the diets is presented in Table 1. All animals received a Revalor S (Intervet GesmbH, Austria) hormone implant at the beginning of the starter phase as per standard feedlot operations in South Africa.
Animals received the starter diet for 21 days, followed by the grower (fed for 80 days) and the finisher (fed for 14 days) diet. Three days was used for adaptation between phases by decreasing the percentage of the former diet and increasing the percentage of the new diet until the animals received only the diet of the new phase. Animals were given ad libitum access to feed and water.
Eight animals, one from each pen, were randomly selected at the start of the trial to collect rumen in the various phases (32 samples). The same animals were used for rumen collection throughout the trial. A trained veterinarian inserted a flexible plastic oesophageal tube into the rumen through the mouth to the ventral sac of the rumen to collect rumen. The microbial community composition of samples collected via oesophageal tube with fluid and solid particles was comparable with those collected via rumen fistula (Paz et aí., 2016). Care was taken to ensure fluid and solid particles were present in the samples. The first 50 ml of rumen content was discarded because of potential saliva contamination. A further 50 ml was collected in a sterilized 50 ml plastic container. Immediately after collection the pH was measured using a portable pH meter (EcoSense pH100A, YSI Environmental, USA) and the sample was frozen in liquid nitrogen and stored at -80 °C until DNA extraction. Owing to technical problems with the portable pH meter, the pH readings of the backgrounding phase had to be discarded.
After thawing, the samples (300 mg) were homogenized using a BeadBug microtube homogenizer (Benchmark Scientific, USA) for approximately 12 mins at maximum speed (400 x 10 rpm) and the DNA was extracted with a QIAamp PowerFecal Pro DNA extraction kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. After extraction, DNA concentration and purity (A260/A280) were quantified with a Qubit fluorometer v2 (Invitrogen, USA) and a Nanodrop ND-1000 spectrophotometer (Thermo Scientific, USA).
Thirty-two DNA samples were shipped to Novogene (NovogeneAIT, Singapore) for pair-ended (250 x 250 bp) sequencing with an Illumina NovaSeq 250 (Illumina, San Diego, California, USA). Sequencing targeted the V3-V4 hypervariable region of the 16S rRNA gene and the 5F region of ITS1. Data were received from Novogene Singapore with primers removed with an average number of reads per sample of 198 775 and 194 585 for 16S rRNA and ITS sequencing. Sequence data was deposited into the NCBI Sequence Read Archive under accession number PRJNA721531.
Microbiome analysis was performed with R software v z4.0.2 (R Core Team, 2013). Processing and analysis of reads were performed using DADA2 v1.16.0 (Callahan et aí., 2016) including read filtering, dereplications, sample inference, chimera removal, merging of paired-end reads, and taxonomic classification. Reads were trimmed at 220 base pairs for both the forward and reverse reads of the 16S rRNA generated reads, resulting in 186 933 reads remaining after trimming. Taxonomy was assigned to genus level with RDP database (Cole et aí., 2014) for 16S rRNA and UNITE database (Nilsson et aí., 2019) for ITS. The ape package v5.4.1 (Paradis & Schliep, 2019) was used to construct a phylogenetic tree. The amplicon sequence variant (ASV) table, taxonomy table, phylogenetic tree and sample data were combined to construct a phyloseq object using phyloseq v1.32.0 (McMurdie & Holmes, 2013). The microbiome package v1.10.0 (Lahti et aí., 2017) was used to generate figures.
Low abundance ASVs (detected at least 10 times in 5% of the samples) were removed for downstream analysis. Reads were rarefied to minimum sampling depth for normalization. The numbers of remaining reads were reported as average and standard deviation. Weighed and unweighted UniFrac distances were used to perform a principle coordinate analysis (PCoA) for ordination analysis to visualize differences between the phases. Beta diversity was determined with Adonis, betadisper and permutest functions in vegan v2.5.6 (Oksanen et aí., 2020). This linear model was used to test for beta diversity with these functions: unifrac.dist ~ phase + animal + group. These same fixed effects (phase, animal, group) were tested for significant influence on alpha diversity and the relative abundance of the microbes with an analysis of variance. Animal and group were found not to have a significant effect and were not included in further tests. Three alpha diversity indices were calculated using phyloseq, namely the observed number of ASVs, Chao1 richness estimator, and Shannon diversity index. Kruskal-Wallis and Wilcoxon rank sum tests were used to determine statistical significance for the relative abundance of the taxa and alpha diversity. Analysis was corrected for multiple testing with Bonferroni correction. For all statistical tests, results were considered significant at P <0.05 and trends were recognized at P <0.1.
Results and Discussion
Following quality control, chimera detection and removal, the samples had an average read count of 116 943 ± 19 832 for the 16S rRNA and 149 447 ± 15 014 for the ITS sequencing. From the sequences, 42 008 and 35 442 amplicon sequence variants (ASVs) were detected for 16S rRNA and ITS sequencing.
There was no difference in the rumen microbiome composition between the control and monensin groups (P >0.05), whereas a difference (P = 0.001) was observed between the four phases for the 16S rRNA and ITS rumen populations.
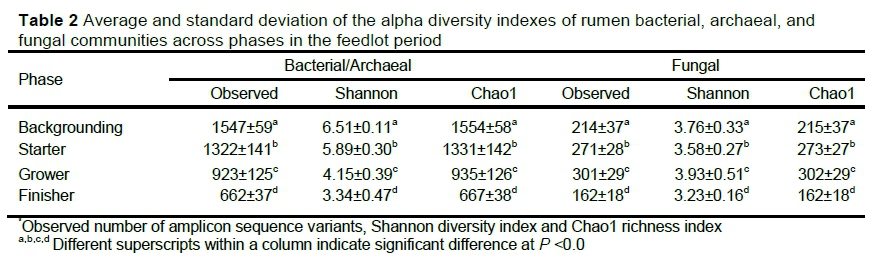
In Table 2, alpha diversity is shown based on the observed number of ASVs, Shannon and Chaol indices. The diversity in the samples was higher for the backgrounding phase, with a consequent decrease up to the finisher phase for the bacterial and archaea population. Most ASVs for the fungi population were observed in the grower period, with the least in the finisher phase. All alpha diversity indices were significantly different across the four phases, with no significant difference between the groups or animals.
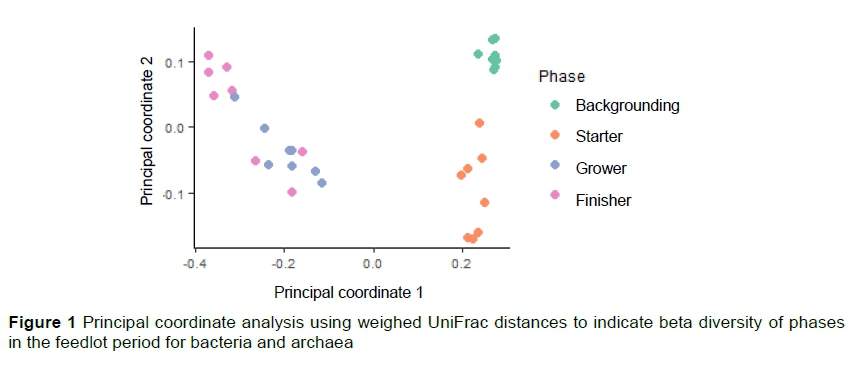
There was a difference in beta diversity between the phases (P = 0.001, R2 = 0.601), but not between the animals or groups (P = 0.395, R2 = 0.301). The backgrounding and starter phases clustered separately (Figure 1), whereas the grower and the finisher phases formed a larger dispersed cluster. The first principle coordinate explained 53.3% of the variation in the diversity of bacteria and archaea whereas the second principle coordinate only explained 7.1%.
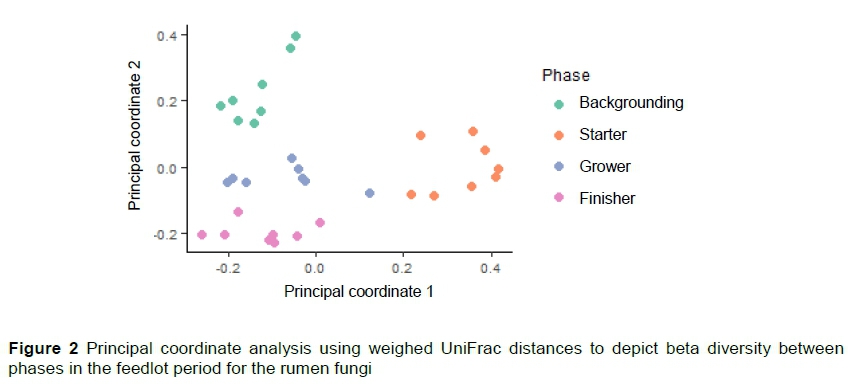
In Figure 2, a PCoA using weighed UniFrac distance of the fungal population of the phases can be observed to cluster separately. The groups (monensin and control) did not cluster separately within the phases. The first principle coordinate explained 32.6% of the variation in the rumen fungi whereas the second principle coordinate explained 20.1%.
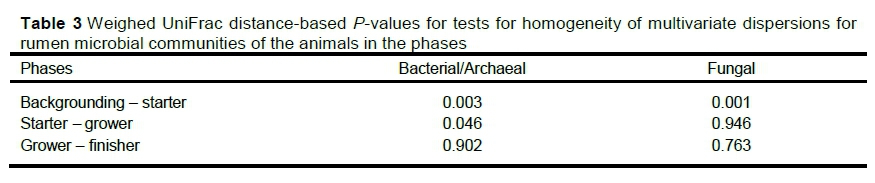
There was a difference in microbial composition between the phases (P =0.001, R2 =0.568), but not between the animals or groups (P =0.221, R2 =0.123) in the fungal population. There was also a difference in dispersion between the phases (P =0.002). The significance tests based on weighed UniFrac distances for the homogeneity of multivariate dispersions are reported in Table 3. There was a significant difference in the microbial composition between the backgrounding and starter phases in the 16S rRNA and between the starter and grower phases as shown in the ordination plot (Figure 1) and confirmed by the permutation test. As seen in Figure 2, the rumen fungi population present during the backgrounding phase clustered separately from the starter phase, showing a significance difference between the communities of these two phases (Table 3).
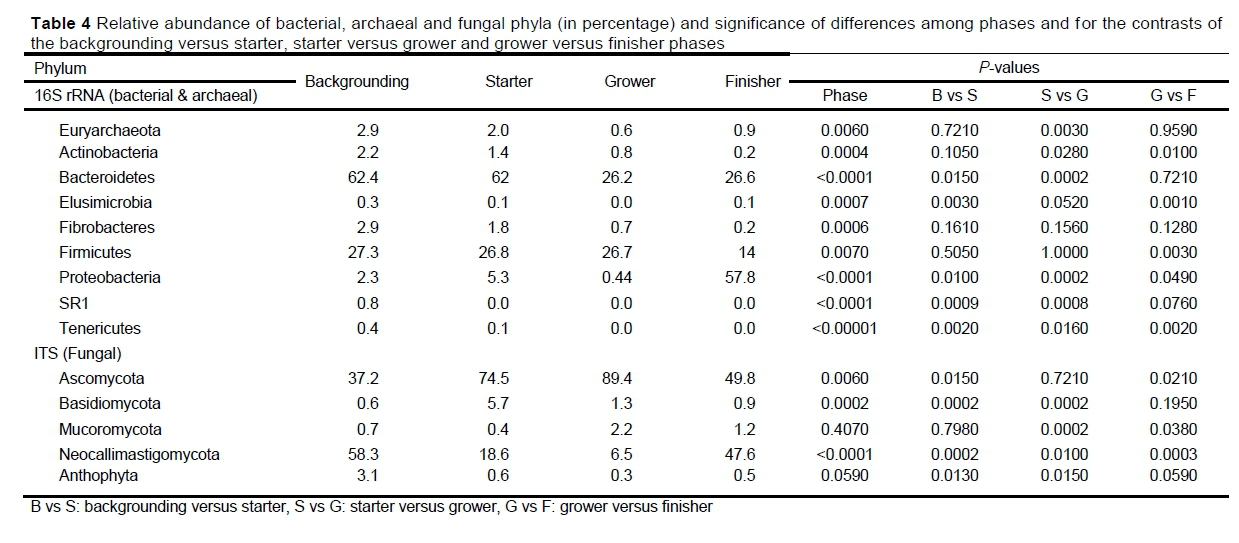
The abundance of the phyla (in percentage) and significance of the various factors are depicted in Table 4. Most phyla did not differ (P >0.05) between the groups or between the animals. Overall, the most abundant phylum in the rumen microbiome was Bacteroidetes (56%), followed by Firmicutes (29%) and Proteobacteria (5%) (Figure 3). At phylum level, 2% of the microbes were not characterized. Of the ASVs identified in the rumen, 0.7% were archaea from the Euryarchaeota phylum. The relative abundance of most phyla did not differ (P >0.05) between the groups or between the animals.
At family level, Prevotellaceae from the Bacteroidetes phylum and Ruminococcaceae from the Firmicutes phylum were the most abundant overall, with Prevotellaceae more abundant in the starter phase, whereas Ruminococcaceae was more abundant in the grower phase (Figure 4). Porphyromonadaceae, Lachnospiraceae, Sphingobacteriaceae, Fibrobacteraceae and Methanobacteriaceae were also present.
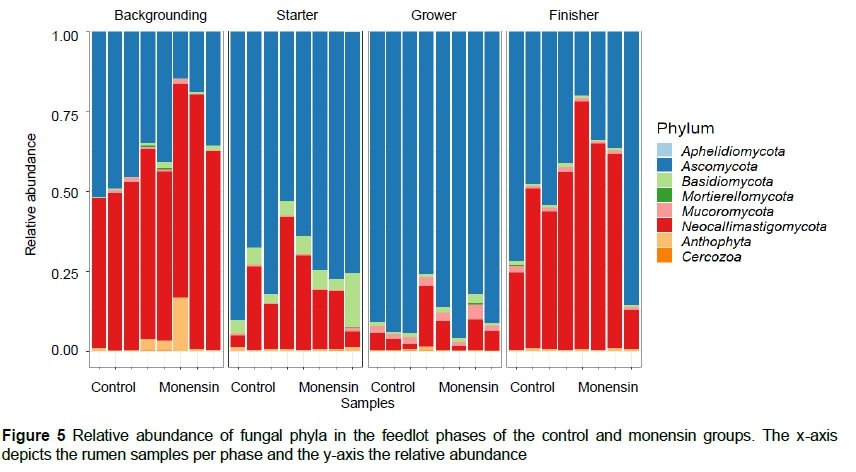
For the fungi population, Ascomycota, Neocallistigomycota and Basidiomycota were the most abundant (Figure 5, Table 4). Ascomycota was more abundant in the grower period, whereas Neocallimastigomycota was more abundant in the finisher phase.
At phylum level, 45% of the ASVs were non-characterized. Neocallimastigaceae from the Neocallimastigomycota phylum and Aspergillaceae from the Ascomycota phylum were the most abundant (Figure 6).
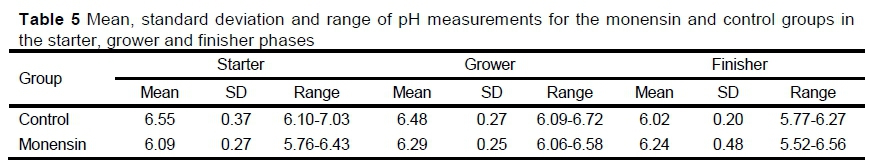
There was no difference (P <0.5) in the pH measurement between the phases or between the control and monensin groups (Table 5).
Microbial diversity in the rumen microbiome determines the amount of energy, in the form of VFAs and other products, available to the animal for production (Guan et al., 2008; Shabat et al., 2016) with a higher propionate to acetate ratio resulting in more energy (Wolin, 1960). The weighed UniFrac distances differentiated bacterial and fungal communities between the phases significantly. This was expected as the composition of the diets differed, and diet is one of the most influential factors on microbial composition (Belanche et al., 2012; Gruninger et al., 2019; Stanton et al., 2020).
Feed additives such as monensin are commonly used in feedlots in South Africa. Monensin is known to decrease the abundance of gram-positive bacteria and methanogens by limiting the nutrient supply, resulting in a decrease in the acetate to propionate ratio and in methane emissions (Boadi et al., 2004; Thomas et al., 2017). This decrease could result in a more energy efficient process. Feed additives have an effect on rumen microbiome composition (Schären et al., 2017). In this study no significant difference was found in bacterial, archaeal and fungal populations for the group receiving monensin and the control. This finding warrants investigation as it may hold positive outcomes for countries or situations in which feed additives are not allowed in feedlot diets.
The most notable change in the rumen microbiome composition occurred during the transition from a forage-based to a concentrate-based diet as fermentation substrates switched from cellulolytic to amylolytic (Carberry et al., 2012). This was observed in this study where the bacterial population of the backgrounding phase differed significantly from the starter phase, with alpha diversity indexes indicating a higher richness in the backgrounding phase. Roughage-based diets have a wider range of carbohydrate substrates such as cellulose and heteropolysaccharides that are fermented by microbes, resulting in a more diverse rumen microbiome (Belanche et al., 2012). These diets have a less acidic rumen environment, which plays a role in rumen diversity because many microbes are sensitive to acidic conditions (Russell & Wilson, 1996). The pH measurements in this study (pH 6.0-7.0) indicated a less acidic environment for the starter phase.
Bacteroidetes phylum was the most abundant during backgrounding, as expected for animals on a roughage diet (Li et al., 2012). Prevoteíía from the Bacteroidetes phylum was the most abundant genus in backgrounding compared with the starter phase. Several studies reported that Prevoteíía was the most abundant bacterial genus in the rumen microbiome, regardless of diet (Stevenson & Weimer, 2007; Jami & Mizrahi, 2012) because it is involved in the degradation of multiple substrates (Rosewarne et al., 2014) and production of acetate, succinate and propionate (Carberry et al., 2012; Chen et al., 2017).
Ruminococcus, Cíostridium and Pseudobutyrivibrio from the Firmicutes phylum are plant fibre degraders (Danielsson et al., 2017) and were expected to be abundant in the backgrounding phase. Pseudobutyrivibrio had a significant effect on average daily feed intake (Paz et al., 2018), and average daily gain (Myer et al., 2015). Methanobrevibacter and Pseudobutyrivibrio were both more abundant in the backgrounding phase compared with the starter phase. These microbes were shown to be correlated because Pseudobutyrivibrio was identified as a potential biomarker for methane emissions (Auffret et al., 2018) and Methanobrevibacter is a methanogen (Tapio et al., 2017). Their lower abundance in the starter phase could be because of the higher energy content of the diet because propionate is favoured over acetate production. This results in more energy being available to the animal for production (Jeyanathan et al., 2019).
Fungi play a role in degrading fibrous materials in the rumen (Gruninger et al., 2014) and are therefore more abundant in roughage-based diets, such as during backgrounding. However, in this study the number of observed ASVs showed a higher abundance of fungi in the starter phase. Further research is needed. Most studies (Gruninger et al., 2014; Zhang et al., 2017, 2020; Belanche et al., 2019) reported that Neocallimastigomycota was the most abundant fungal phylum in the rumen microbiome. In contrast, the Ascomycota phylum was the most abundant fungus in the current study, followed by Basidiomycota and Neocallimastigomycota. Few studies are available for Ascomycota and Basidiomycota as these phyla are aerobic fungi (Zhang et al., 2020) and are rarely found in animals (Zhang et al., 2017). Zhang et al. (2017) reported an increase in their abundance as the proportion of concentrates increased in dairy cattle, which was also observed in this study. It is unclear how these aerobic microbes survived in the anaerobic environment of the rumen. However, they might play a role in scavenging oxygen entering the rumen and have a beneficial effect on the anaerobic fermentation in the rumen (Zhang et al., 2020).
Aspergillaceae family from the Ascomycota phylum have been used as feed additives in animal nutrition (Adegbeye et al., 2020) because they decrease methane emissions by reducing the growth and activity of methanogenic bacteria (Wolin & Miller, 2006). The abundance of Aspergillus in the starter phase might indicate a decrease in methane emissions. The decrease in Aspergiííusis from the backgrounding phase to the starter phase is in line with the earlier observations regarding Methanobrevibacter and Pseudobutyrivibrio.
There is a beneficial symbiotic relationship between anaerobic fungi from the Neocallimastigomycota phylum and methanogens, such as Methanobrevibacter (Cheng et al., 2009). Methanobrevibacter and the Neocallistigaceae family were more abundant in the backgrounding phase. Roughage-based diets have a higher methane production per unit of feed compared with diets high in concentrates (Beauchemin & McGinn, 2006).
he most prominent shift in the rumen microbiome composition was between the starter and the grower phases. The increase in the proportion of carbohydrates in the diet shifts the rumen microbial composition from predominantly Firmicutes to Proteobacteria (Petri et al., 2018). The proportion of carbohydrates in the diet has a significant effect on the rumen microbiome population (Raabis et al., 2019) as an increase in easily digested carbohydrates results in more propionate-producing bacteria, lower fibre-degrading organisms, lower protein breakdown, and higher feed efficiency (Fernando et al., 2010; Belanche et al., 2012). Bacteroidetes was the most abundant phylum in the starter phase, whereas Proteobacteria was more abundant in the grower phase. Microbes from the Proteobacteria phylum have diverse metabolic functions and indicate an increase in the number of bacteria that are metabolically capable of handling easily fermentable carbohydrates (Fernando et al., 2010). Succinivibrio was more abundant in the starter phase, whereas Vampirovibrio and Ruminobacter were more abundant in the grower phase. A higher abundance of propionate-producing bacteria such as Succinivibrio (Suen et al., 2011) may divert hydrogen from methanogens, thus reducing enteric methane emissions and increasing available energy for metabolism (De Menezes et al., 2011).
Eubacterium and Ruminococcus showed significantly higher abundance in the grower phase. Eubacterium is abundant in efficient steers and has a tolerance of low pH, whereas Ruminococcus has been associated with residual feed intake (Hernandez-Sanabria et al., 2012). Because the grower diet had a higher proportion of easily fermentable carbohydrates, a lower pH could be expected in comparison with the starter phase. The pH measurements in this study did not show the expected decrease in pH from starter to grower. This might be explained by possible saliva contamination in the samples because of the method of rumen collection or selective feeding by the cattle before collection. In highly efficient steers, the acetate utilization characteristics of Eubacterium may interact with the acetate-producing capacity of Succinivibrio to utilize excessive hydrogen, which otherwise would be directed to methanogenesis (Chassard & Bernalier-Donadille, 2006).
Based on alpha diversity, more fungi were present in the grower phase compared with the starter. This was unexpected, as fungi are known to decrease in abundance as the proportion of carbohydrates in the diet increase. A low pH, which would be found in high concentrate diets such as the grower diet, can inhibit the growth of anaerobic fungi (Han et al., 2019), leading to a decrease in their abundance. Cyllamyces and Orpinomyces were the genera more abundant in the starter phase. These genera from the Neocallistigomycota phylum are known to be present in the rumen (Gruninger et al., 2014; Zhang et al., 2017) and degrade cellulose and xylose (Kittelmann et al., 2012). Genera from Ascomycota (Neoascochyta, Selenophoma and Cecomyces) were more abundant in the grower phase compared with the starter. However, few studies in the literature elucidated their abundance in the starter phase. Belanche et al. (2019) reported that fungi could be ingested with feed materials such as plant pathogens, saprotrophs, yeast and other species of unclassified fungi. The abundance of these genera might be because of external factors and their role in the rumen requires further research to confirm their origin and functions.
There was no significant difference in the rumen microbe population between the grower and finisher groups for either the bacteria and archaea or the fungi population. This might be because Proteobacteria had a high abundance in both the grower and finisher phases. Even though the high abundance of Proteobacteria could indicate possible dysbiosis in the rumen of the cattle (Auffret et al., 2017), no physical effects of acidosis or metabolic disorders were observed in the animals during the grower and finisher phases. Many pathogenic bacteria belong to the Proteobacteria phylum and these pathogens are sensitive to dietary change (Baümler & Sperandio, 2016). Metabolic diseases in cattle such as bloat or acidosis have been associated with an unbalanced rumen microbiome (Khafipour et al., 2009) and are known to occur in finishing diets. High concentrate diets, such as the finisher diet in this study, increase production of lactate and are associated with acid tolerant microbes such as Proteobacteria (Fernando et al., 2010). Because the finisher period had the highest abundance of Proteobacteria, strategies could be formulated to decrease the abundance of pathogenic microbes while maintaining beneficial microbes.
The finisher phase had the lowest alpha diversity compared with the other phases. A lower alpha diversity has been associated with more efficient animals (Zhou et al., 2009; Shabat et al., 2016). It is therefore more desirable to have low diversity in the rumen microbial population to focus on promoting energy yield from the feed (Shabat et al., 2016). However, a too low alpha diversity has been associated with an unbalanced and unhealthy rumen microbiome composition. It is therefore imperative to balance the microbiome composition to prevent dysbiosis.
The finisher phase exhibited the least abundance of fungi compared with the other phases based on the alpha diversity. A similar observation was reported by Kumar et al. (2015), but was in contrast to Zhang et al. (2017). This decrease in the richness and diversity of the fungi in the finisher might be because of the higher proportion of concentrates as fungi are mostly fibre degrading microbes. Neocaííismastix from the Neocallistogomycota phylum was the most abundant in the finisher phase. This genus is able to utilize a wide range of substrates such as cellulose, xylose, glucose, starch, grass, and straw (Edwards et al., 2017). However, its abundance decreased with increasing concentrates (Han et al., 2019), which is in contrast with this study. Further studies on the function and prevalence of fungi in the rumen microbiome are needed.
There are other factors that must be mentioned when discussing the rumen microbiome. Although most of the differences discussed above can be explained by the influence of diet and the age of the animals (Jami et al., 2013), host genetics can influence the rumen microbiome. Studies reported that animals fed the same diet could exhibit substantial differences in microbiome composition (Welkie et al., 2010; Firkins & Yu, 2015) because of host genetics and the interaction between the host and the rumen microbiome (Hernandez-Sanabria et al., 2013).
Conclusion
This is the first study to investigate the rumen microbiome of South African Bonsmara cattle under intensive feedlot conditions. Improvement of feed efficiency in feedlot cattle holds several advantages, including cost, a decrease in environmental impact and food safety. There was no significant difference in the overall rumen microbiome population between the monensin and the control groups, but differences within the phases require further investigation. This study allowed for an improved understanding of microbial shift in the feedlot period. This understanding could provide integrative information about rumen function and lead to improved ruminant production through changes in digestibility and feed efficiency.
Acknowledgements
The authors would like to thank Ms Tanita Botha for her advice on the statistical analysis, Red Meat Research and Development South Africa and Sernick Group (PTY) LTD for funding the project and the Meat Industry Trust for the bursary to support DAL.
Authors' contributions
All authors participated in the planning of the project. DAL conducted the trial, performed the laboratory work, bioinformatics and statistical analysis and wrote the original paper. EvMK supervised the project, wrote, and edited the paper. CJLdT assisted in the nutritional aspects of the project. DS assisted in the bioinformatic and statistical analysis. mMs revised the paper. All authors have read and agreed to the published version of the manuscript.
Conflict of interest declaration
The authors declare there is no conflict of interest.
References
Adegbeye, M.J., Kanth, P.R., Obaisi, A.I., Elghandour, M.M.M.Y., Oyebamiji, K.J., Salem, A.Z M., Morakinyo-Fasipe, O.T. & Cipriano-Salazar, M., 2020. Sustainable agriculture options for production, greenhouse gasses and pollution alleviation, and nutrient recycling in emerging and transitional nations - An overview. J. Clean. Prod. 242, 118319. https://doi.org/10.1016/j.jclepro.2019.118319 [ Links ]
Auffret, M.D., Dewhurst, R.J., Duthie, C., Rooke, J.A., Wallace, R.J., Freeman, T.C., Stewart, R., Watson, M.& Roehe, R., 2017. The rumen microbiome as a reservoir of antimicrobial resistance and pathogenicity genes is directly affected by diet in beef cattle. Microbiome 5, 159. https://doi.org/10.1186/s40168-017-0378-z [ Links ]
Auffret, M.D., Stewart, R., Dewhurst, R.J., Duthie, C., Rooke, J.A., Wallace, R.J., Freeman, T.C., Snelling, T.J., Watson, M. & Roehe, R., 2018. Identification, comparison, and validation of robust rumen microbial biomarkers for methane emissions using diverse Bos taurus breeds and basal diets. Front. Microbiol. 8, 1-15. https://doi.org/10.3389/fmicb.2017.02642 [ Links ]
Baümler, A.J. & Sperandio, V., 2016. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535, 85-93. https://doi.org/10.1038/nature18849 [ Links ]
Beauchemin, K.A.& McGinn, S.M., 2006. Methane emissions from beef cattle: Effects of fumaric acid, essential oil, and canola oil. J. Anim. Sci. 84, 1489-1496. https://doi.org/10.2527/2006.8461489x [ Links ]
Belanche, A., Kingston-Smith, A.H., Griffith, G.W. & Newbold, C.J., 2019. A multi-kingdom study reveals the plasticity of the rumen microbiota in response to a shift from non-grazing to grazing diets in sheep. Front. Microbiol. 10. https://doi.org/10.3389/fmicb.2019.00122 [ Links ]
Belanche, A., Doreau, M., Edwards, J.E., Moorby, J.M., Pinloche, E. & Newbold, C.J., 2012. Shifts in the rumen microbiota due to the type of carbohydrate and level of protein ingested by dairy cattle are associated with changes in rumen fermentation. J. Nutr. 142, 1684-1692. https://doi.org/10.3945/jn.112.159574 [ Links ]
Bevans, D.W., Beauchemin, K.A., Schwartzkopf-Genswein, K.S., McKinnon, J.J. & McAllister, T.A., 2005. Effect of rapid or gradual grain adaptation on subacute acidosis and feed intake by feedlot cattle. J. Anim. Sci. 83, 1116-1132. https://doi.org/10.2527/2005.8351116x [ Links ]
Boadi, D.A., Wittenberg, K.M., Scott, S.L., Burton, D., Buckley, K., Small, J.A. & Ominski, K.H., 2004. Effect of low and high forage diet on enteric and manure pack greenhouse gas emissions from a feedlot. Can. J. Anim. Sci. 445453. https://cdnsciencepub.com/doi/pdf/10.4141/A03-079 [ Links ]
Callahan, B.J., McMurdie, P.J., Rosen, M.J., Han, A.W., Johnson, A.J.A. & Holmes, S.P., 2016. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581-583. https://doi.org/10.1038/nmeth.3869 [ Links ]
Capper, J.L., 2011. Replacing rose-tinted spectacles with a high-powered microscope: The historical versus modern carbon footprint of animal agriculture. Anim. Front. 1, 26-32. https://doi.org/10.2527/af.2011-0009 [ Links ]
Carberry, C.A., Kenny, D.A.D.A., Han, S., McCabe, M.S.M.S. & Waters, S.M.S.M., 2012. The effect of phenotypic residual feed intake (RFI) and dietary forage content on the rumen microbial community of beef cattle. Appl. Environ. Microbiol. 1-42. https://doi.org/10.1128/AEM.07759-1 [ Links ]
Chassard, C. & Bernalier-Donadille, A., 2006. H2 and acetate transfers during xylan fermentation between a butyrate- producing xylanolytic species and hydrogenotrophic microorganisms from the human gut. FEMS Microbiol. Lett. 254, 116-122. https://doi.org/10.1111/j.1574-6968.2005.00016.x [ Links ]
Chen, T., Long, W., Zhang, C., Liu, S., Zhao, L. & Hamaker, B.R., 2017. Fiber-utilizing capacity varies in Prevotella- versus ßactero/des-dominated gut microbiota. Sci. Rep. 7, 2594. https://doi.org/10.1038/s41598-017-02995-4 [ Links ]
Cheng, Y.F., Edwards, J.E., Allison, G.G., Zhu, W.Y. & Theodorou, M.K., 2009. Diversity and activity of enriched ruminal cultures of anaerobic fungi and methanogens grown together on lignocellulose in consecutive batch culture. Bioresour. Technol. 100, 4821-4828. https://doi.org/10.1016/j.biortech.2009.04.031 [ Links ]
Cole, J. R., Wang, Q., Fish, J.A., Chai, B., McGarrell, D.M., Sun, Y., Brown, C.T., Porras-Alfaro, A., Kuske, C.R. & Tiedje, J.M., 2014. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, 633-642. https://doi.org/10.1093/nar/gkt1244 [ Links ]
DAFF (Department of Agriculture, Forestry and Fisheries), 2019. A profile of the South African beef market value chain. https://www.dalrrd.gov.za/doaDev/sideMenu/Marketing/Annual%20Publications/Be ef%20Market%20Value%20Chain%20Profile%202019.pdf [ Links ]
Danielsson, R., Dicksved, J., Sun, L., Gonda, H., Müller, B., Schnürer, A. & Bertilsson, J., 2017. Methane production in dairy cows correlates with rumen methanogenic and bacterial community structure. Front. Microbiol. 8, 1 -15. https://doi.org/10.3389/fmicb.2017.00226 [ Links ]
De Menezes, A.B., Lewis, E., O'Donovan, M., O'Neill, B.F., Clipson, N. & Doyle, E.M., 2011. Microbiome analysis of dairy cows fed pasture or total mixed ration diets. FEMS Microbiol Ecol 78, 256-265. https://doi.org/10.1111/j.1574-6941.2011.01151.x [ Links ]
Edwards, J.E., Forster, R.J., Callaghan, T.M., Dollhofer, V., Dagar, S.S., Cheng, Y., Chang, J., Kittelmann, S., Fliegerova, K., Puniya, A.K., Henske, J.K., Gilmore, S.P., O'Malley, M.A., Griffith, G.W. & Smidt, H., 2017. PCR and omics based techniques to study the diversity, ecology and biology of anaerobic fungi: Insights, challenges and opportunities. Front. Microbiol. 8. https://doi.org/10.3389/fmicb.2017.01657 [ Links ]
Fernando, S.C., Purvis, H.T., Najar, F.Z., Sukharnikov, L.O., Krehbiel, C.R., Nagaraja, T.G., Roe, B.A. & DeSilva, U., 2010. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 76, 7482-7490 https://doi.org/10.1128/AEM.00388-10 [ Links ]
Firkins, J.L. & Yu, Z., 2015. Ruminant Nutrition Symposium: How to use data on the rumen microbiome to improve our understanding of ruminant nutrition. J. Anim. Sci. 93, 1450-1470. https://doi.org/10.2527/jas.2014-8754. [ Links ]
Gruninger, R.J., Puniya, A.K., Callaghan, T.M., Edwards, J.E., Youssef, N., Dagar, S., Fliegerova, K., Griffith, G.W., Forster, R., Tsang, A., Mcallister, T. & Elshahed, M.S., 2014. Anaerobic fungi (phylum Neocallimastigomycota): Advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS Microbiol. Ecol. 90, 1-17. https://doi.org/10.1111/1574-6941.12383 [ Links ]
Gruninger, R.J., Ribeiro, G.O., Cameron, A. & McAllister, T.A., 2019. Invited review: Application of meta-omics to understand the dynamic nature of the rumen microbiome and how it responds to diet in ruminants. Animal 13(9), 1843-1854. DOI: 10.1017/S1751731119000752 [ Links ]
Guan, L.L., Nkrumah, J.D., Basarab, J.A. & Moore, S.S., 2008. Linkage of microbial ecology to phenotype: Correlation of rumen microbial ecology to cattle's feed efficiency. FEMS Microbiol. Lett. 288, 85-91. https://doi.org/10.1111/j.1574-6968.2008.01343.x [ Links ]
Han, X., Li, B., Wang, X., Chen, Y. & Yang, Y., 2019. Effect of dietary concentrate to forage ratios on ruminal bacterial and anaerobic fungal populations of cashmere goats. Anaerobe 59, 118-125. https://doi.org/10.1016/j.anaerobe.2019.06.010 [ Links ]
Hernandez-Sanabria, E., Goonewardene, L.A., Wang, Z., Durunna, O.N., Moore, S.S. & Guan, L.L., 2012. Impact of feed efficiency and diet on adaptive variations in the bacterial community in the rumen fluid of cattle. Appl. Environ. Microbiol. 78, 1203-1214. https://doi.org/10.1128/AEM.05114-11 [ Links ]
Hernandez-Sanabria, E., Goonewardene, L.A., Wang, Z., Zhou, M., Moore, S.S. & Guan, L.L., 2013. Influence of sire breed on the interplay among rumen microbial populations inhabiting the rumen liquid of the progeny in beef cattle. PLoS One 8. https://doi.org/10.1371/journal.pone.0058461 [ Links ]
Huws, S.A., Creevey, C.J., Oyama, L.B. & Mizrahi, I., 2018. Addressing global ruminant agricultural challenges through understanding the rumen microbiome: Past, present, and future. Front. Microbiol. 9, 1 -33. https://doi.org/10.3389/fmicb.2018.02161 [ Links ]
Jami, E. & Mizrahi, I., 2012. Composition and similarity of bovine rumen microbiota across individual animals. PLoS One 7, 1-8. https://doi.org/10.1371/journal.pone.0033306 [ Links ]
Jami, E., Israel, A., Kotser, A. & Mizrahi, I., 2013. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 7, 1069-1079. https://doi.org/10.1038/ismej.2013.2 [ Links ]
Jeyanathan, J., Martin, C., Eugène, M., Ferlay, A., Popova, M. & Morgavi, D.P., 2019. Bacterial direct-fed microbials fail to reduce methane emissions in primiparous lactating dairy cows. J. Anim. Sci. Biotechnol. 10, 1-9. https://doi.org/10.1186/s40104-019-0342-9 [ Links ]
Khafipour, E., Li, S., Plaizier, J.C. & Krause, D. O., 2009. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl. Environ. Microbiol. 75, 7115-7124. https://doi.org/10.1128/AEM.00739-09 [ Links ]
Kittelmann, S., Naylor, G.E., Koolaard, J.P. & Janssen, P.H., 2012. A proposed taxonomy of anaerobic fungi (class Neocallimastigomycetes) suitable for large-scale sequence-based community structure analysis. PLoS One 7, 113. https://doi.org/10.1371/journal.pone.0036866 [ Links ]
Klieve, A.V., Hennessy, D., Ouwerkerk, D., Forster, R.J., Mackie, R.I. & Attwood, G.T., 2003. Establishing populations of Megasphaera eísdenii YE 34 and Butyrivibrio fibrisoívens YE 44 in the rumen of cattle fed high grain diets. J. Appl. Microbiol. 95, 621-630 https://doi.org/10.1046/j.1365-2672.2003.02024.x [ Links ]
Koenig, K.M., Chibisa, G.E., Penner, G.B. & Beauchemin, K.A., 2020. Optimum roughage proportion in barley-based feedlot cattle diets: growth performance, feeding behavior, and carcass traits. J. Anim. Sci. 98, 1-12. https://doi.org/10.1093/jas/skaa299 [ Links ]
Kumar, S., Indugu, N., Vecchiarelli, B. & Pitta, D. W., 2015. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front. Microbiol. 6, 1-10. https://doi.org/10.3389/fmicb.2015.00781 [ Links ]
Lahti, L. & Shetty, S. et al., 2017. Tools for microbiome analysis in R. https://microbiome.github.io/tutorials/ [ Links ]
Li, R.W., Connor, E.E., Li, C., Baldwin, R.L. & Sparks, M.E., 2012. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 14, 129-139. https://doi.org/10.1111/j.1462-2920.2011.02543.x [ Links ]
Mackie, R.I., Gilchrist, F.M.C., Robberts, A.M., Hannah, P.E. & Schwartz, H.M. 1978. Microbiological and chemical changes in the rumen during the stepwise adaptation of sheep to high concentrate diets. J. Agric. Sci. 90, 241-254. https://doi.org/10.1017/S0021859600055313. [ Links ]
McMurdie, P.J. & Holmes, S., 2013. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. https://doi.org/10.1371/journal.pone.0061217 [ Links ]
Myer, P.R., 2019. Bovine genome-microbiome interactions: Metagenomic frontier for the selection of efficient productivity in cattle systems. mSystems 4, e00103-19. https://journals.asm.org/doi/10.1128/mSystems.00103-19 [ Links ]
Myer, P.R., Smith, T.P.L.L., Wells, J.E., Kuehn, L.A. & Freetly, H.C., 2015. Rumen microbiome from steers differing in feed efficiency. PLoS One 10, 1-17. https://doi.org/10.1371/journal.pone.0129174 [ Links ]
Nilsson, R.H., Larsson, K.H., Taylor, A.F.S., Bengtsson-Palme, J., Jeppesen, T.S., Schigel, D., Kennedy, P., Picard, K., Glöckner, F.O., Tedersoo, L., Saar, I., Kõljalg, U. & Abarenkov, K., 2019. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259- D264. https://doi.org/10.1093/nar/gky1022 [ Links ]
Oksanen, J., Blanchet, F.G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., Minchin, P.R., O'Hara, R.B., Simpson, G.L., Solymos, P., Stevens, M. H., Szoecs, E. & Wagner, H., 2020. vegan: R package for community ecology. https://cran.r-project.org, https://github.com/vegandevs/vegan [ Links ]
Paradis, E. & Schliep, K., 2019. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526-528. https://doi.org/10.1093/bioinformatics/bty633 [ Links ]
Paz, H.A., Anderson, C.L., Muller, M.J., Kononoff, P.J. & Fernando, S.C., 2016. Rumen bacterial community composition in Holstein and Jersey cows is different under same dietary condition and is not affected by sampling method. Front. Microbiol. 7, 1-9. https://doi.org/10.3389/fmicb.2016.01206 [ Links ]
Paz, H.A., Hales, K.E., Wells, J.E., Kuehn, L.A., Freetly, H.C., Berry, E.D., Flythe, M.D., Spangler, M.L. & Fernando, S.C., 2018. Rumen bacterial community structure impacts feed efficiency in beef cattle. J. Anim. Sci. 96, 1045-1058. https://doi.org/10.1093/jas/skx081 [ Links ]
Perea, K., Perz, K., Olivo, S.K., Williams, A., Lachman, M., Ishaq, S.L., Thomson, J. & Yeoman, C.J., 2017. Feed efficiency phenotypes in lambs involve changes in ruminal, colonic, and small-intestine-located microbiota. J. Anim. Sci. 95, 2585-2592. https://doi.org/10.2527/jas2016.1222 [ Links ]
Petri, R.M., Kleefisch, M.T., Metzler-Zebeli, B.U., Zebeli, Q. & Klevenhusen, F., 2018. Changes in the rumen epithelial microbiota of cattle and host gene expression in response to alterations in dietary carbohydrate composition. Appl. Environ. Microbiol. 84, 1-14. https://doi.org/10.1128/AEM.00384-18 [ Links ]
Pitta, D.W., Indugu, N., Baker, L., Vecchiarelli, B. & Attwood, G., 2018. Symposium review: Understanding diet - microbe interactions to enhance productivity of dairy cows. J. Dairy Sci. 101, 7661-7679. https://doi.org/10.3168/jds.2017-13858 [ Links ]
R Core team, 2013. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing [ Links ]
Raabis, S., Li, W. & Cersosimo, L., 2019. Effects and immune responses of probiotic treatment in ruminants. Vet. Immunol. Immunopathol. 208, 58-66. https://doi.org/10.1016/j.vetimm.2018.12.006 [ Links ]
Rosewarne, C.P., Pope, P.B., Cheung, J.L. & Morrison, M., 2014. Analysis of the bovine rumen microbiome reveals a diversity of Sus-like polysaccharide utilization loci from the bacterial phylum Bacteroidetes. J. Ind. Microbiol. Biotechnol. 41, 601-606. https://doi.org/10.1007/s10295-013-1395-y [ Links ]
Russell, J.B. & Wilson, D.B. 1996. Why are ruminal cellulolytic bacteria unable to digest cellulose at low pH? J. Dairy Sci. 79, 1503-1509. https://doi.org/10.3168/jds.S0022-0302(96)76510-4 [ Links ]
Samuelson, K.L., Hubbert, M.E., Galyean, M.L. & Löest, C. A., 2016. Nutritional recommendations of feedlot consulting nutritionists: The 2015 New Mexico State and Texas Tech University Survey. J. Anim. Sci. 94, 2648-2663. https://pubmed.ncbi.nlm.nih.gov/27285940/ [ Links ]
Schären, M., Kiri, K., Riede, S., Gardener, M., Meyer, U., Hummel, J., Urich, T., Breves, G., Dänicke, S. & Jami, E., 2017. Alterations in the rumen liquid-, particle- and epithelium-associated microbiota of dairy cows during the transition from a silage- and concentrate-based ration to pasture in spring. Front. Microbiol. 8, 744. https://doi.org/10.3389/fmicb.2017.00744 [ Links ]
Shabat, S.K. Ben, Sasson, G., Doron-Faigenboim, A., Durman, T., Yaacoby, S., Miller, M.E.B., White, B.A., Shterzer, N. & Mizrahi, I., 2016. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 10, 2958-2972 https://doi.org/10.1038/ismej.2016.62 [ Links ]
Stanton, C., Leahy, S., Kelly, B., Ross, R.P. & Attwood, G., 2020. Manipulating the rumen microbiome to address challenges facing Australasian dairy farming. Anim. Prod. 60, 36-45. https://agris.fao.org/agris-search/search.do?recordID=US202000297699 [ Links ]
Stevenson, D.M.& Weimer, P.J., 2007. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Environ. Microbiol. 75, 165-174. https://doi.org/10.1007/s00253-006-0802-y [ Links ]
Suen, G., Stevenson, D.M., Bruce, D.C., Chertkov, O., Copeland, A., Cheng, J.-F., Detter, C., Detter, J.C., Goodwin, L.A., Han, C.S., Hauser, L.J., Ivanova, N.N., Kyrpides, N.C., Land, M.L., Lapidus, A., Lucas, S., Ovchinnikova, G., Pitluck, S., Tapia, R., Woyke, T., Boyum, J., Mead, D. & Weimer, P.J., 2011. Complete genome of the cellulolytic ruminal bacterium Ruminococcus albus 7. J. Bacteriol. 193, 5574-5575. https://doi.org/10.1128/JB.05621-11 [ Links ]
Tajima, K., Aminov, R.I., Nagamine, T., Matsui, H., Nakamura, M. & Benno, Y., 2001. Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl. Environ. Microbiol. 67, 2766-74. https://doi.org/10.1128/AEM.67.6.2766-2774.2001 [ Links ]
Tapio, I., Snelling, T.J., Strozzi, F.& Wallace, R.J., 2017. The ruminal microbiome associated with methane emissions from ruminant livestock. J. Anim. Sci. Biotechnol. 1 -11. https://doi.org/10.1186/s40104-017-0141-0 [ Links ]
Thomas, M., Webb, M., Ghimire, S., Blair, A., Olson, K., Fenske, G.J., Fonder, A.T., Christopher-Hennings, J., Brake, D. & Scaria, J., 2017. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci. Rep. 7, 1-13. https://doi.org/10.1038/s41598-017-12481-6. [ Links ]
Welkie, D.G., Stevenson, D.M. & Weimer, P.J., 2010. ARISA analysis of ruminal bacterial community dynamics in lactating dairy cows during the feeding cycle. Anaerobe 16, 94-100. https://doi.org/10.1016/j.anaerobe.2009.07.002 [ Links ]
Wolin, M.J. 1960. A theoretical rumen fermentation balance. J. Dairy Sci. 43, 1452-1459. DOI: 10.3168/jds.S0022- 0302(60)90348-9 [ Links ]
Wolin, M.J. & Miller, T.L., 2006. Control of rumen methanogenesis by inhibiting the growth and activity of methanogens with hydroxymethylglutaryl-SCoA inhibitors. International Congress Series 1293, 131-137. https://doi.org/10.1016/j.ics.2006.01.031 [ Links ]
Zhang, Y., Li, F., Chen, Y., Wu, H. & Meng, Q., 2020. Metatranscriptomic profiling reveals the effect of breed on active rumen eukaryotic composition in beef cattle with varied feed efficiency. Front. Microbiol. 11, 1-12. https://doi.org/10.3389/fmicb.2020.00367 [ Links ]
Zhang, J., Shi, H., Wang, Y., Li, S., Cao, Z., Ji, S., He, Y. & Zhang, H., 2017. Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in Holstein heifers. Front. Microbiol. 8, 1 -18. https://doi.org/10.3389/fmicb.2017.02206 [ Links ]
Zhou, M., Hernandez-Sanabria, E. & Guan, L.L.2009. Assessment of the microbial ecology of ruminal methanogens in cattle with different feed efficiencies. Appl. Environ. Microbiol. 75, 6524-6533. https://doi.org/10.1128/AEM.02815-08 [ Links ]
Submitted 10 November 2021
Accepted 24 December 2021
Published 9 April 2022
# Corresponding author: u11084210@tuks.co.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}