










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Animal Science
On-line version ISSN 2221-4062
Print version ISSN 0375-1589
S. Afr. j. anim. sci. vol.50 n.3 Pretoria 2020
http://dx.doi.org/10.4314/sajas.v50i3.9
ARTICLES
Effects of supplementing geese with green sweet sorghum stalks on microbiota in segments of the gastrointestinal tract
H. Zhong; Z. Liu; M. Liang; Q. Wang; Y. Wang; Y. Luo; J. Sun; C. Zhang; Q. Li; C. Wang#
Chongqing Academy of Animal Sciences, Chongqing, China
ABSTRACT
To explore the effects of supplementing a basal diet with green sweet sorghum stalks on the gut microbiota in geese, one hundred and twenty 28-day-old geese were divided into two groups. Group 1 was fed a basal diet and group 2 was fed 92% of the same basal diet with green sweet sorghum stalks ad libitum for 42 days. Three male geese from each group were killed at 70 days old, four gastrointestinal tract segments (duodenum, jejunum, ileum, and caecum) were collected, and the hypervariable V4 region of the bacterial 16S rRNA gene was sequenced. There were obvious changes of microbiome in the caecum compared with the other three intestinal segments after green sweet sorghum stalks were added to the diet. Group 2 had significantly higher alpha diversity in the caecum compared with group 1 (P <0.05). The phyla of bacterial communities in the caecum differed. Group 2 had more abundant Bacteroidetes and Firmicutes, but Proteobacteria were more abundant in group 1. At genus level, Bacteroides was more prevalent in group 2, as were Prevotella and YRC22 (P <0.05). Functional analysis revealed that the carbohydrate metabolism, membrane transport, endocrine system and digestive system metabolic pathways were overrepresented in group 2. In conclusion, caecal microbes might play an important role in digesting green sweet sorghum stalks in geese.
Keywords: caecum, goose, microbiome, microbial diversity
Introduction
In 2017 global animal feed volume was estimated at 1.07 billion metric tons with a grossed industrial output value of about 430 million dollars (Johansen, 2018). In the same year, industrial feed production exceeded 0.2 billion metric tons in China, making it the largest producer of feed in the world for seven consecutive years. However, competition between animals and people for food has increased gradually to the extent that it has become a primary concern for food security (Liu et al., 2018). The development and use of unconventional feed resources are important to alleviate this problem and to reduce the cost of food for animals (Song et al., 2015, Ülger et al., 2017,Ülger et al., 2018).
Sweet sorghum (Sorghum bicolor (L.) Moench) is a domesticated species the stalks of which have high levels of juice and edible sugar (Murray et al., 2009). As a typical unconventional feedstuff, sweet sorghum stalks have been used successfully to feed pigs (Passos et al., 2015), cattle (Amer et al., 2010), and sheep (Anandan et al., 2012). Sweet sorghum stalks could be added to diets for poultry. A previous study investigated the possibility of utilizing green sweet sorghum stalk (henceforth 'sorghum') diets for geese between 28 and 70 days old (Huang et al., 2017). In that study, the control group was provided a basal diet ad libitum and five other groups were fed 96%, 92%, 88%, 84%, and 80% of this basal diet, with sorghum being provided ad libitum. It was concluded that 4% to 8% of the basal diet could be replaced with sorghum without influencing growth, carcass yield, and meat quality of Sichuan white geese. The treatment in group 2 proved the most effective compromise between feed cost and performance, suggesting that geese could derive nutritional value from sorghum stalks. Average daily consumption of sorghum was 74.83 g, whereas consumption of the basal diet was reduced from 191.1 g to 180.2 g per day.
Although geese have no cellulose-digesting enzymes in their digestive tract, they are able to utilize plant-derived coarse fodder, and thus are considered a good model in which to study the function of intestinal microbiome. Previous research has studied the effects on microbial diversity in the caecum of adding cassava foliage (Li et al., 2017), ryegrass and Chinese trumpet creeper (Xu et al., 2017) and cornstalk (Liu et al., 2018) to the diets of geese. The results of these studies suggested that diet could affect the composition of gut microbiota. However, the impact of dietary supplementation with sorghum on the gut microbiota of geese has not been studied. Importantly, resection of the caecum significantly reduced digestibility of crude fibre from soybean meal and cottonseed meal in caecectomized geese (Jiang et al., 2013). Based on this result, it was speculated that feeding a basal diet supplemented with sorghum would not greatly influence the microbiota in duodenum, jejunum, and ileum, but would affect the caecal microbiota. Therefore, in this study, geese fed the basal diet and those fed 92% of the same diet supplemented with sorghum ad libitum were hypothesized to have similar populations in their intestinal tracts. The data were generated using Illumina sequencing on the hypervariable V4 region of the bacterial 16S rRNA gene. These data might identify differences in the intestinal microbiome of geese and help in understanding the molecular mechanism of their digestion of sorghum.
Material and Methods
The animal experiment was accomplished in strict accordance with the guidelines for the Care and Use of Experimental Animals established by the Ministry of Science and Technology of the People's Republic of China (Approval number 2006-398). Animal feeding and sampling were approved by the Animal Care and Welfare Committee and the Laboratory Animal Management Committee of Chongqing Academy of Animal Sciences. All efforts were made to reduce the suffering of geese. All animal experiments were carried out at a waterfowl breeding base in Rongchang County, Chongqing City, China.
Apparent nutrient digestibility of sorghum was determined using the total faecal collection method (Liu et al., 2019). Ten Sichuan white geese (male, 60 weeks old) with 4093 g bodyweight were housed singly in metabolic cages that had been cleaned and disinfected. The steel frame metabolic cages were equipped with grid floors and collection trays. Four days of adjustment were allowed prior to a five-day collection period. The whole fresh sorghum was mowed, chopped to 1 - 3 cm in size, and fed to the geese ad libitum. Drinking water was freely available at all times. The collected droppings were weighed and oven-dried at 75 °C. The crude protein content of the dried samples was determined as prescribed by the AOAC (2000). The apparent metabolizable energy content of the sorghum was 10.69 MJkg-1 and the apparent metabolic rate of crude protein was 62.57%.
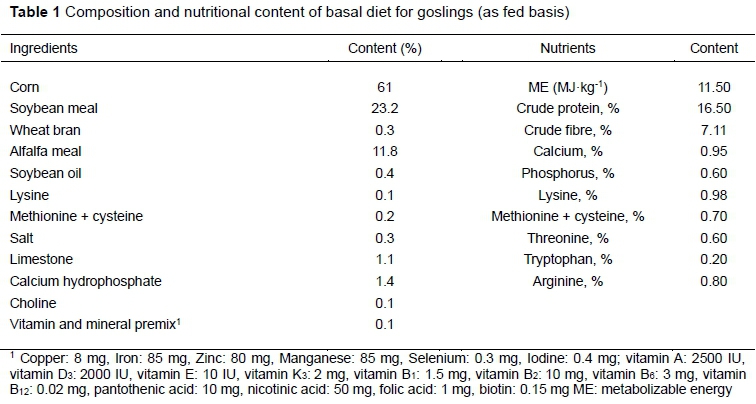
One hundred and twenty 28-day-old Sichuan white goslings weighing approximately 1100 g were randomly allocated to two groups with six replicates of ten birds each. The pens in which they were housed had one feeder and five drinkers. The birds in the first group were provided the basal diet ad libitum (group 1), whereas the birds in the second group were fed 92% of this basal with sorghum ad libitum (group 2). Birds were kept in plastic-wire floored pens in an environmentally controlled goose house. The room temperature was maintained at 20 - 22 °C and with 16 hours of light and 8 hours of darkness. Drinking water was freely available at all times. The feeding period was 42 days. Two-hundred-gram samples of sorghum were collected daily, sealed in hermetic bags, and stored at 4 °C for subsequent nutritional analysis. The gross energy of the sorghum used in this study was 17.58 MJkg-1, and the contents of crude protein, crude fibre, ether extract, and ash were 11.79%, 34.13%, 1.45%, and 3.72%, respectively. The contents of phosphorus, calcium, neutral detergent fibre, acid detergent fibre, and nitrogen-free extract were 0.31%, 0.41%, 64.64%, 37.90%, and 38.54%, respectively. The ingredients of the basal diet are provided in Table 1. At 70 days old, three male geese with bodyweights approximately equal to those of their cohorts were selected from each group and slaughtered. The average bodyweights in groups 1 and 2 were 3200 g and 3130 g, respectively. The content of four intestinal segments, including the duodenum, jejunum, ileum, and caecum, were collected simultaneously under aseptic conditions. All samples were snap-frozen in liquid nitrogen and stored at -80 °C until DNA extraction. Microbial genomic DNA was extracted from the contents of four intestinal segments using the QIAamp DNA stool mini kit (QIAGEN, Hilden, Germany) according to the manufacturer's protocols. The V4 hypervariable regions of 16S rRNA were amplified through polymerase chain reaction (PCR) using the barcoded fusion primers (520F 5'-AYTGGGYDTAAAGNG-3' and 802R 5'-TACNVGGGTATCTAATCC- 3'), as described by Zhao et al. (2013). The amplifications were examined on a 2% agarose gel, and the band was extracted and purified with the AxyPrep DNA gel extraction kit (Corning Inc., Corning, New York, USA) according to the manufacturer's instructions.
The PCR products were used to construct a sequencing library with Illumina TruSeq Nano DNA LT library prep kit (Illumina, Inc., San Diego, California, USA). For each sample, barcoded V4 PCR amplications were sequenced with the Illumina Miseq platform. The DNA extraction, PCR amplification, and sequencing service were accomplished with Personal Biotechnology Co., Ltd. (Shanghai, China).
Sequence reads were eliminated if they included ambiguous bases, if the average Phred score was 25, if a homopolymer ran > 6, if there were mismatches in the primer sequence, and if a sequence length was < 100 bp. Trimmed and assembled sequences were uploaded to QIIME, v1.8.0 (Caporaso et al., 2010).
Bacterial operation taxonomie units (OTUs) with greater than 97% similarity were clustered, and taxonomie identification was assigned using the RDP classifier with a confidence threshold of 0.8 (Wang et al., 2007). Rarefaction analysis based on MOTHUR v1.30.1 was conducted to calculate the chaol and ACE, diversity estimates, and the Shannon and Simpson index-based measures of community evenness (Schloss et al., 2009). To investigate the differences of microbial communities between group 1 and group 2, beta diversity analysis was conducted by using principal coordinate analysis (PCA).
The functional composition of the caecal metagenome was predicted using PICRUSt (Langille et al., 2013). The OTUs at 97% similarity were mapped to gg13.5 database by QIIME, v1.8.0. The abundance of OTUs was normalized automatically using 16S rRNA gene copy numbers from known bacterial genomes in the Integrated Microbial Genomes and Microbiomes (IMG/M) database (Chen et al., 2019) The predicted genes and their function were aligned to the KEGG database (Kanehisa & Goto, 2000) and the differences among groups were compared with the STAMP software (Parks et al., 2010). Student's two-tailed t-test and ANOVA analysis were performed with SPSS 10.0 software (SPSS Inc., Chicago, Illinois, USA) with P <0.05 being considered statistically significant.
Results and Discussion
Research into the microbial diversity in the gastrointestinal tract of domestic poultry has intensified in recent years. To the authors' knowledge, this is the first study to evaluate the effect on their intestinal microbiomes of adding sorghum to the diet fed to geese using high-throughput sequencing.
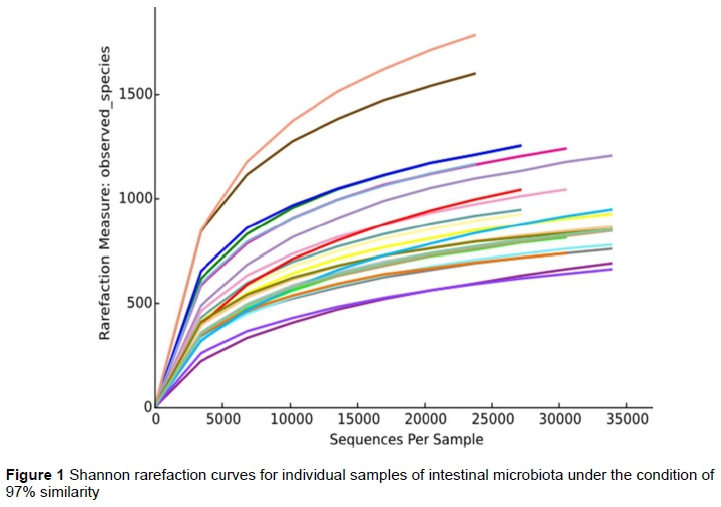
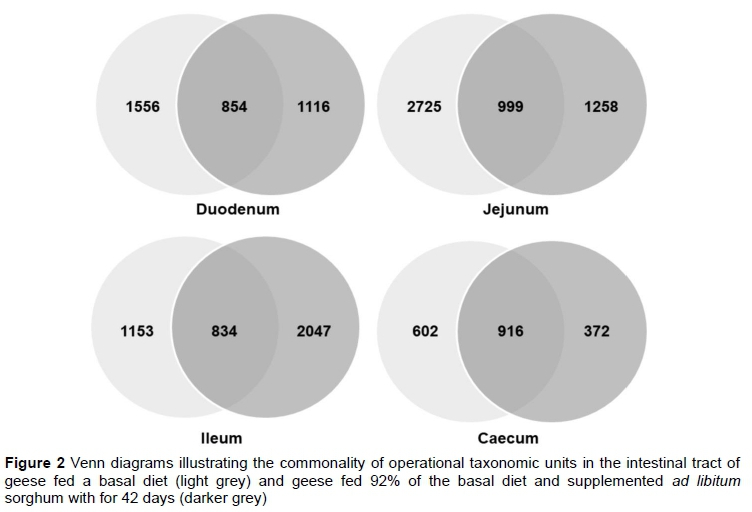
A total of 86 819 qualified sequences were obtained from the duodenum, jejunum, ileum, and caecum of the six geese. There was an average of 36 034 reads per sample. Shannon value and Shannon rarefaction curves for each sample are shown in Figure 1 and indicate that the sequence coverage was adequate. Of the 4201 and 3963 OTUs found for groups 1 and 2, 3398 were shared. In both groups, fewer OTUs were found in the caecal samples than in the other segments of the intestinal tracts (Figure 2).
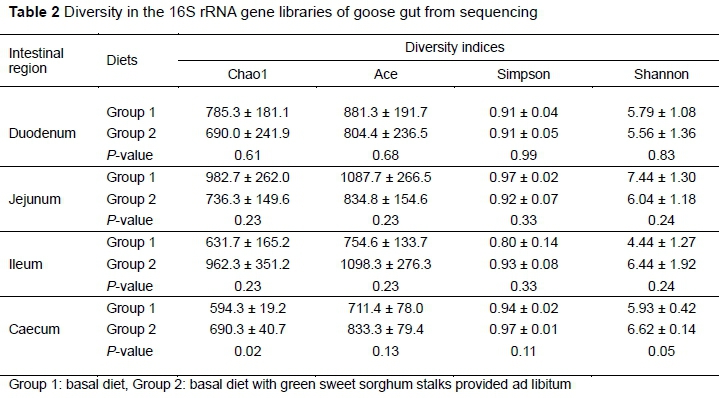
No significant difference in microbial richness (chaol and ACE indices) and evenness (Shannon and Simpson indices) were found in the duodenum, jejunum, and ileum (P >0.05) (Table 2). However, the choa1 index indicated greater richness (P =0.02) of the microbiota in the caecum of geese in group 2. The Shannon index also indicated greater (P =0.05) microbial diversity in these samples.
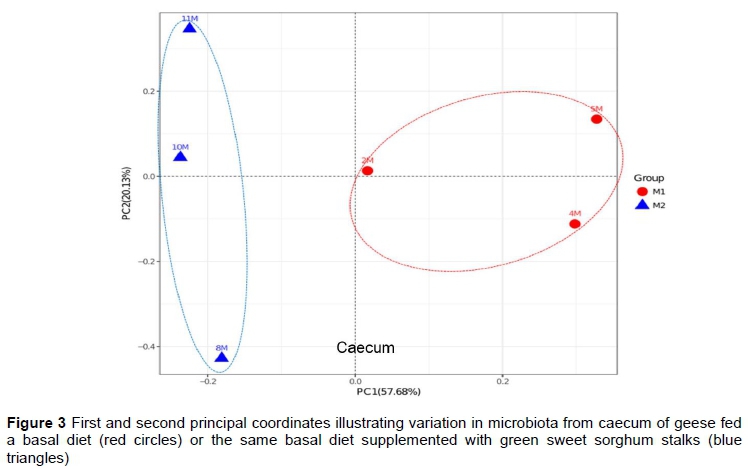
Principal coordinate analysis (R Core Team, 2017) was used to study the community composition in four intestinal segments of the two groups. An unweighted implementation of the UniFrac method proposed by Lozupone and Knight (2005) identified distinct differences in the caecum microbiota from geese fed the two diets, but showed little differentiation in the duodenum, jejunum, and ileum samples. The first and second principal coordinates accounted for 57.68% and 20.13% of the variation between groups in the microbiota of the caecum, respectively (Figure 3). After sorghum was added to the diet, obvious changes in the microbiome were found in the caecum than in the other three intestinal segments of the geese. This finding was similar to results obtained in pigs in which different distributions of microbiota were observed across segments of the gastrointestinal tract (Zhao et al., 2015). Based on the metagenome profiles of adult pigs, they showed enrichment associated with metabolism pathways and metabolic disease in large intestine while higher abundance of infectious disease, immune function disease, and cancer functions were seen in the small intestine. Some scholars believe that dietary fibres are not digested in the small intestine but could be fermented in the large intestine (Macfarlane et al., 2008). The role of the small intestine is thought to be in digesting and absorbing the food from the stomach, with the large intestine and the caecum in particular facilitating microbial fermentation (Dibaise et al., 2008). It is widely accepted that even if they are genetically unable to produce cellulose-digesting enzymes in the intestinal tract, geese possess a strong capability to digest fibrous plant material through microbial fermentation in the caecum (He et al., 2015; Zhou et al., 2015; Xu et al., 2017).
Resection of the caecum significantly reduces the digestibility of crude fibre in soybean meal and cottonseed meal by caecectomized geese (Jiang et al., 2013). Therefore, the authors speculated that feeding 92% of the basal diet supplemented ad libitum with sorghum was not a sufficient dietary alteration to influence the microbiota in duodenum, jejunum and ileum, but that it would have a great effect on the caecal microbiota (Figure 3). More than 90.0% of these sequences belonged to the three most abundant phyla of bacteria, namely Bacteroidetes, Firmicutes and Proteobacteria, in both groups of geese. This is consistent with previous studies (Gao et al., 2016; Li et al., 2017). In musk oxen, the relative abundant of Bacteroidetes and Firmicutes could reach 90% in the rumen gut microbiome and account for most of fibre digestion (Ungerfeld et al., 2018). Bacteroidetes have the ability to degrade polysaccharides and metabolic intermediates (Louis et al., 2007; Lin et al., 2011). Both Firmicutes and Bacteroidetes can produce short-chain fatty acids via fermentation of fat and fibre in human beings (Duncan et al., 2007; Filippo et al., 2010). In group 2, the Firmicutes were numerically more abundant than in group 1 (Table 3). Parnell et al. (2012) observed that prebiotic fibres could increase the amount of Firmicutes and promote the growth of beneficial bacteria in the gut. However, the increasing level of Firmicutes did not improve the energy metabolism in this study.
Synergistetes are anaerobic gram-negative bacteria and opportunistic pathogens. However, Synergistetes can be found in the microbiome of the umbilicus and in the normal vaginal flora of healthy people (Jumasbilak et al., 2009; Marchandin et al., 2010). Synergistetes was observed only in group 2 and their role in fibre digestion remains unknown.
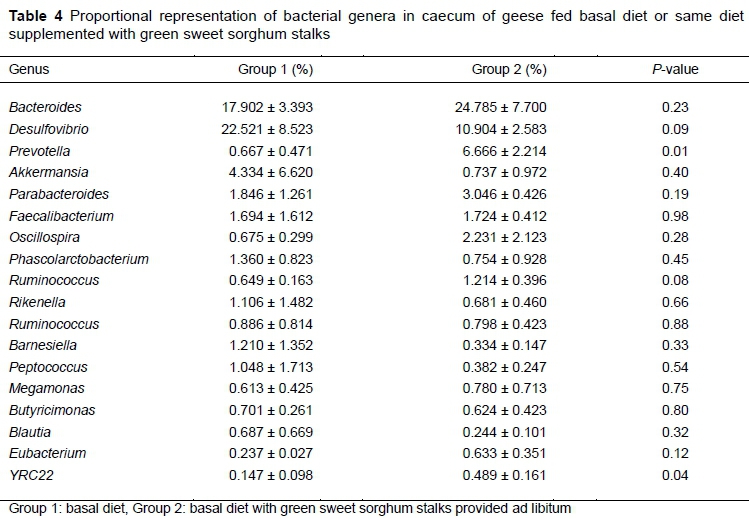
Only 30 OTUs could be identified to genus level (Table 4). Of these, 17 were Firmicutes, 7 were Bacteroidetes, 2 were Proteobacteria, 1 was Verrucomicrobia, 1 was Deferribacteres, 1 was Actinobacteria and 1 was Euryarchaeota. Only one bacterial genus, namely Methanocorpusculum (0.283%), was present in the caecal samples of group 2, whereas it was absent in group 1. Bacteroides and Desulfovibrio were predominant genera that represented 40.423% and 35.689% in group 1 and group 2, respectively. Only two OTUs, Prevotella and YRC22, were significantly different in abundance between geese on the two diets (P =0.010 and P =0.035, respectively). The relative abundance of Prevotella in group 2 was nearly nine times that of group 1. Similarly, the relative abundance of YRC22 in the group 2 was approximately three times greater than in group 1. Methanocorpusculum was present in the caecal samples from group 2 in low frequency (0.28%) but was almost absent from group 1.
Linear discriminant analysis effect size from QIIME indicated overrepresentation of seven genera in the caecal microbiome of geese in group 2, and five overrepresented genera in geese fed the basal diet (Figure 4).
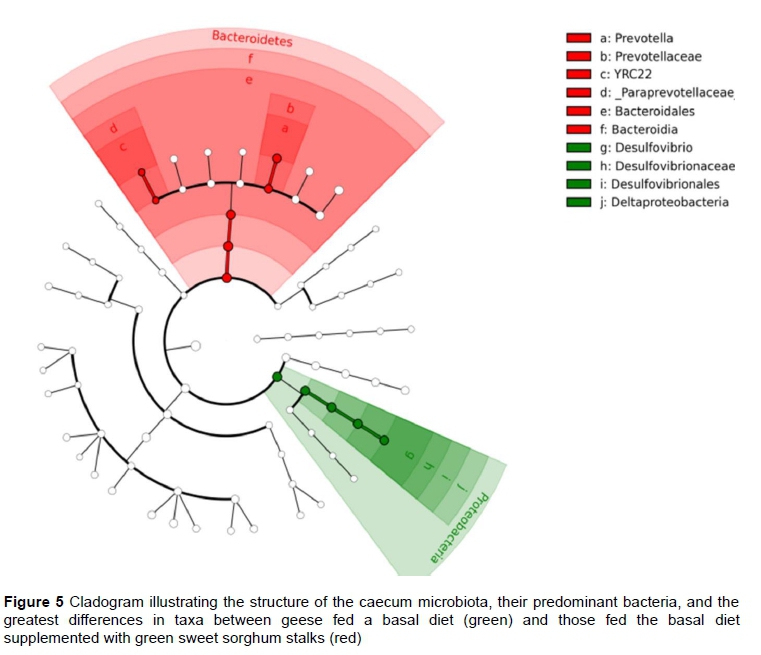
The cladogram represents the structure of the caecum microbiota, their predominant bacteria, and the greatest differences in taxa between the groups (Figure 5), Two genera (Prevotella, YRC22), two families (Prevotellaceae, Paraprevotellaceae), and one order (Bacteroidales) belonging to the phylum Bacteroidetes were heavily weighted in group 2. One genus (Desulfovibrio), one family (Desulfovibrionaceas), one order (Desulfovibrionales), and one class (Deltaproteobacteria) belonging to the phylum Proteobacteria were heavily weighted in group 1.
Taxa abundance of Prevotella and YRC22 in the two groups were compared using Metastats analysis (White et al., 2009) (Figure 6). The results suggest that Prevotella and YRC22 were more abundant in the caecum of geese whose diet was supplemented with sorghum. group 2.
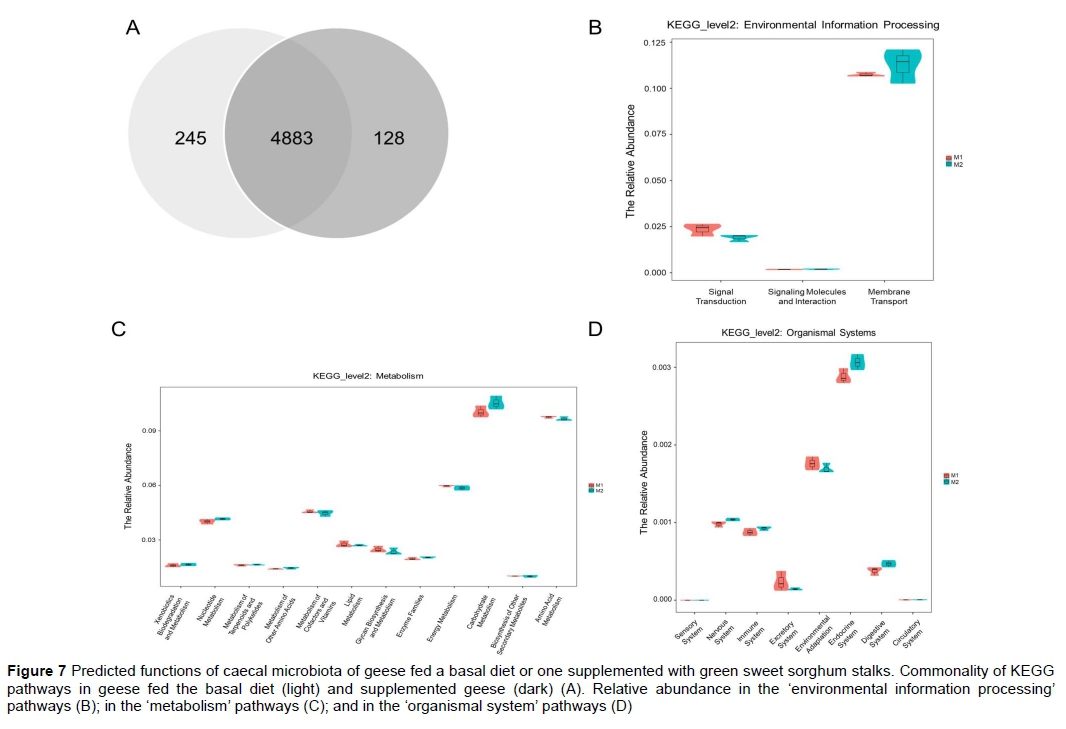
The OTUs were normalized by 16S rRNA gene copy number and their metagenomic functions predicted from KEGG pathways. A total of 5256 KEGG pathways were found, of which 245 were specific to group 1, 128 were unique to group 2, and 4883 were shared (Figure 7A). Unfortunately, few OTUs (only 1 to 27) were represented in group 2-specific pathways. Intriguingly, the KEGG orthology (KO) of carbohydrate metabolism, which belonged to 'metabolism', membrane transport, which belonged to 'environmental information processing', and endocrine system and digestive system, which belonged to 'organismal system', were overrepresented in group 2 relative to group 1 (Figure 7B, 7C, 7D). Second, further analysis of the KEGG pathways was conducted based on the 20 top-ranked differentially represented OTUs (Table 5). Of these, four KOs did not match the KEGG pathways (K02004, K02003, K07024, and K07090). Of the other 16 KOs, eight were more represented in group 2, namely K06147, K02025, K02026, K00266, K02529, K02006, K09687, and K01190, which are involved in membrane transport and carbohydrate metabolism.
The genera Prevotella and Bacteroides are well-known fermenters of dietary fibre. Their abundance in the caecum of geese in group 1 was 18.6%. However, with the addition of the sorghum, the proportion of Prevotella and Bacteroides was 31.5%. Prevotella in the caecum can ferment carbohydrates, thus supplying short-chain fatty acids, which are important for host health (Macfarlane et al., 1995). Prevotella are also able to degrade the mucin glycoprotein and improve intestinal conditions to promote the growth and survival of the host (Dp et al., 2000; Rho et al., 2005). During the postweaning period, the high-level abundance of Prevotella in pigs has been related to degradation of hemicelluloses, including xylans (Hayashi et al., 2007; Lamendella et al., 2011). A recent study showed that dietary supplementation with an organic acid could lead to an increased abundance of Prevotella, which facilitated degradation of carbohydrates and thereby strengthened intestinal immunity (Li et al., 2018).
The level of dietary fibre affects the intestinal microflora of goose (Zhou et al., 2018). Geese fed a lower level of dietary fibre (5% corn stover or alfalfa) had fewer (P <0.05) Prevotella in the caecal microflora than those that were fed a diet that was higher in fibre (8% corn stover or alfalfa). The present findings are consistent with this result and explain the relationship between dietary fibre and the abundance of Prevotella in the caecal microflora of geese.
The genus YRC22 was significantly more prevalent in the caecum in group 2 than in group 1 (P <0.05). However, little is currently known about YRC22 and its function, although it has been found in the caecal microbiota of birds and mammals (Venable et al., 2016).
Conclusions
This study provides a foundation for understanding the utilization of sorghum by goose gut microbiota. Supplementation with chopped sorghum could affect caecal microbiota favourably by increasing the abundance of genera Prevotella and YRC22. Promotion of the genera Prevotella andYRC22, together with Bacteroides, might enhance carbohydrate metabolic, membrane transport, endocrine system and digestive system functions in goose caecum. However, fibre digestion, absorption and utilization by the gut microflora in geese is a complicated process, and further research is required to uncover the underlying mechanisms.
Acknowledgments
This work was supported by The National Key Research and Development Program of China (2018YFD0501505); Chongqing Performance Incentive and Guide Special Projects (cstc2017jxjl80041 or internal number: 17425); the Earmarked Fund for China Agriculture Research System (CARS-42-22); the Basic and Frontier Research Program of Chongqing (No. cstc2016jcyjA0092). Sequencing service was provided by Personal Biotechnology Co., Ltd. Shanghai, China.
Authors' Contributions
HZ, ZL and ML contributed equally to this work. CW conceived and designed the experiments. ZL, YL, CZ, ML, QL and YW conducted the field trial, and collected the samples. HZ and JS wrote the paper. QW and CW discussed and reviewed the paper.
Conflict of Interest Declaration
The authors have no conflict of interest to declare.
References
Amer, S. & Mustafa, A.F., 2010. Short communication: Effects of feeding pearl millet silage on milk production of lactating dairy cows. J. Dairy Sci. 93(12), 5921-5925. [ Links ]
Anandan, S., Zoltan, H., Khan, A.A., Ravi, D. & Blümmel, M., 2012. Feeding value of sweet sorghum bagasse and leaf residues after juice extraction for bio-ethanol production fed to sheep as complete rations in diverse physical forms. Anim. Feed Sci. Tech. 175(3), 131-136. [ Links ]
AOAC, 2000.Official methods of analysis. 17th edition. AOAC International, Rockville, Maryland, USA. [ Links ]
Caporaso, J., Kuczynski, J., Stombaugh, J., Bittinger, K. & Bushman, F., 2010. QIIME allows integration and analysis of high-throughput community sequencing data. Nat. Meth. 7(5) 335-336. [ Links ]
Chen, I.A., Chu, K., Palaniappan, K., Pillay, M., Ratner, A., Huang, J., Huntemann, M., Varghese, N., White, J.R., Seshadri, R., Smirnova, T., Kirton, E., Jungbluth, S.P., Woyke, T., Eloe-Fadrosh, E.A., Ivanova, N.N., & Kyrpides, N.C., 2019. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res., 47(D1), D666-D677. https://doi.org/10.1093/nar/gky901 [ Links ]
Dibaise, J.K., Zhang, H., Crowell, M.D., Krajmalnik-Brown R., Decker, G.A. & Rittmann, B.E., 2008. Gut microbiota and Its possible relationship with obesity. Mayo Clinic Proceedings 83(4), 460-469. [ Links ]
Dp, W., Rosendale, D.I. & Robertson, A.M., 2000. Prevotella enzymes involved in mucin oligosaccharide degradation and evidence for a small operon of genes expressed during growth on mucin. Fems Microbiol. Letters 190(2000), 73-79. [ Links ]
De Filippo, C. Cavalieri, D., Paola, M.D., Ramazzotti, M., Poullet, J.B., Massart, S., Collini, S., Pieraccini G. & Lionetti, P., 2010. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Nat. Acad. Sci. USA 107(33), 14691-14696. [ Links ]
Duncan, S.H., Louis, P. & Flint, H.., 2007. Cultivable bacterial diversity from the human colon. Lett. Appl. Microbiol. 44(4), 343-350. [ Links ]
Gao, G., Zhao, X., Li, Q., He, C., Zhao, W., Liu, S., Ding, J., Ye, W., Wang, J. & Chen Y., 2016. Genome and metagenome analyses reveal adaptive evolution of the host and interaction with the gut microbiota in the goose. Sci. Rept. 6, 32961. [ Links ]
Hayashi, H., Shibata, K.S., M, Tomita, S. & Benno, Y., 2007. Prevotella copri sp. nov. and Prevotella stercorea sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 57(5), 941-946. [ Links ]
He, L.W., Meng, Q.X., Li, D.Y., Zhang, Y.W. & Ren, L.P., 2015. Influence of feeding alternative fiber sources on the gastrointestinal fermentation, digestive enzyme activities and mucosa morphology of growing Greylag geese. Poult. Sci. 94(10), 2464-2471. [ Links ]
Huang, Y., Ma, J.L., Wang, Q.G., Dong, G.Z., Xie, M., Hou, S.S., Liu, Z.L. & Wang, C., 2017. A substitution effect of green sweet sorghum stalks for diet on the growth performance, carcass yields and meat quality in geese. Chinese J. Anim. Vet. Sci. 48(3), 483-491. [ Links ]
Jiang,Y., Wang, Z., Yang, H. & Zhou, C., 2013. Effect of caecectomy on the fiber and protein digestibility of five common feedstuffs. Anim. Husb. Vet. Med. 45(10), 9-12. [ Links ]
Johansen, J., 2018., 2018 Alltech global feed survey results. http://agwired.com/2018/01/25/2017-alltech-global-feed-survey-results/ [ Links ]
Jumasbilak, E., Roudière, L. & Marchandin, H., 2009. Description of 'Synergistetes' phyl. nov. and emended description of the phylum 'Deferribacteres' and of the family Syntrophomonadaceae, phylum 'Firmicutes'. Intern. J. Syst. Evol. Microbiol. 59(5), 1028-1035. [ Links ]
Kanehisa, M. & Goto, S., 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes, Nucleic Acids Res.28(1), 27 -30. https://doi.org/10.1093/nar/28.L27 [ Links ]
Lamendella, R., Domingo, J.W.S., Ghosh, S., Martinson, J. & Oerther, D.B., 2011. Comparative fecal metagenomicsunveils unique functional capacity of the swine gut. BMC Microbiol. 11(1), 103-119. [ Links ]
Langille, M.G.I., Zaneveld, J., Caporaso, J.G., McDonald, D., Dan, K., Reyes, J.A., Clemente, J.C., Burkepile, D.E., Thurber, R.L.V. & Knight R., 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotech. 31(9), 814-821. [ Links ]
Li, M., Zhou, H., Pan, X., Xu, T., Zhang, Z., Zi, X. & Jiang, Y., 2017. Corrigendum: Cassava foliage affects the microbial diversity of Chinese indigenous geese caecum using 16S rRNA sequencing. Sci. Rept. 7, 46837. [ Links ]
Li, S., Zheng, J., Deng, K., Chen, L., Zhao, X.L., Jiang, X.M., Fang, Z.F., Che, L.Q., Xu, S.Y. & Feng, B., 2018. Supplementation with organic acids showing different effects on growth performance, gut morphology and microbiota of weaned pigs fed with highly or less digestible diets. J. Anim. Sci. 96(8). 3302-3318. [ Links ]
Lin, B., Gong, J., Wang, Q., Cui,S., Yu, H. & Huang, B., 2011. In-vitro assessment of the effects of dietary fibers on microbial fermentation and communities from large intestinal digesta of pigs. Food Hydrocolloids 25(2), 180-188. [ Links ]
Liu, G., Luo, X., Zhao, X., Zhang, A., Jiang, N., Yang, L., Huang, M., Xu, L., Ding, L., Li, M., Guo, Z., Li, X., Sun J., Zhou, J., Feng, Y., He, H., Wu, H., Fu, X. & Meng H., 2018. Gut microbiota correlates with fiber and apparent nutrients digestion in goose. Poult. Sci. 97(11), 3899-3909. [ Links ]
Liu, Y., Xiong, K., Yang, S., Liu, X., Luo, Z. & Yang, Y.U., 2018.Research in the application of hay in healthy feeding of cattle and sheep. China Feed 4,13-18. [ Links ]
Liu, Z., Huang, X., Luo, Y., Xue, J., Wang, Q. & Wang, Y., 2019. Effect of dry and wet feed on growth performance, carcass traits, and apparent nutrient digestibility in geese. J. Appl. Poult. Res. 0,1-6. [ Links ]
Louis, P. & Flint, H.J., 2007. Development of a semiquantitative degenerate real-time pcr-based assay for estimation of numbers of butyryl-coenzyme A (CoA) CoA transferase genes in complex bacterial samples. Appl. Environ. Microbiol. 73(6), 2009-2012. [ Links ]
Lozupone, C. & Knight, R., 2005. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71(12), 8228-8235. DOI: 10.1128/AEM.71.12.8228-8235.2005 [ Links ]
Macfarlane, G.T. & Gibson, G. 1995. Microbiological aspects of short chain fatty acid production in the large bowel. Cambridge University Press, Cambridge, England. [ Links ]
Macfarlane, G.T., Steed, H. & Macfarlane S., 2008. Bacterial metabolism and health-related effects of galacto-oligosaccharides and other prebiotics. J. Appl. Microbiol. 104(2), 305-344. [ Links ]
Marchandin, H., Damay, A., Roudière, L., Teyssier, C., Zorgniotti, I., Dechaud, H., Jean-Pierre, H. & Jumas-Bilak, E., 2010. Phylogeny, diversity and host specialization in the phylum Synergistetes with emphasis on strains and clones of human origin. Res. Microbiol. 161(2), 91-100. [ Links ]
Minoru Kanehisa, M. & Goto, S., 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27-30. https://doi.org/10.1093/nar/28.L27 [ Links ]
Murray, S.C., Rooney, W.L., Hamblin, M.T., Mitchell, S.E. & Kresovich, S. 2009. Sweet sorghum genetic diversity and association mapping for brix and height. Plant Genome 2(1), 48-62. [ Links ]
Parks, D.H. & Beiko, R.G., 2010. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26(6), 715-721. [ Links ]
Parnell, J.A. & Reimer, R.A., 2012. Prebiotic fiber modulation of the gut microbiota improves risk factors for obesity and the metabolic syndrome. Gut Microbes 3(1), 29-34. [ Links ]
Passos, A.A., Andrade, C., Phillips, C.E., Coffey, M.T. & Kim, S.W., 2015. Nutrient value of spray field forages fed to pigs and the use of feed enzymes to enhance nutrient digestibility. J. Anim. Sci. 93(4), 1721-1728. [ Links ]
R Core Team, 2017. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ [ Links ]
Rho, J., Wright, D.P., Christie D.L., Clinch, K., Furneaux, R.H. & Roberton, A.M., 2005. A novel mechanism for desulfation of mucin: Identification and cloning of a mucin-desulfating Glycosidase (Sulfoglycosidase) from Prevotella strain RS2. J. Bacteriol. 187(5), 1543-1551. [ Links ]
Schloss, P.D., Westcott, S.L., Ryabin T., Hall, J.R., Hartmann, M., Hollister, E.B., Lesniewski, R.A., Oakley, B.B., Parks, D.H. & Robinson, C.J., 2009. Introducing Mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75(23), 7537. [ Links ]
Song, Z.T., Wang, H.L., Han, C.W., Cai, R.X., Tian, Y. & Yang, R., 2015. Development and utilization of unconventional feed resources for swine in China. Chinese J. Anim. Sci. 51(10), 62-71. [ Links ]
Ungerfeld, E.M., Leigh, M.B., Forster, R.J. & Barboza, P.S., 2018. Influence of season and diet on fiber digestion and bacterial community structure in the rumen of muskoxen (Ovibos moschatus). Microorganisms 6(3), 89-104. [ Links ]
Venable, E., Fenton, K., Braner, V., Reddington, C., Halpin, M., Heitz, S., Francis, J., Gulson, N., Goyer, C. & Bland, S., 2016. Effects of feeding management on the equine caecal microbiota. J. Equine Vet. Sci. 49, 113-121. [ Links ]
Wang, Q., Garrity, G.M., Tiedje, J.M. & Cole, J.R., 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73(16), 5261-5267. [ Links ]
White, J.R., Nagarajan, N. & Pop, M., 2009. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. pLoS Comput. Biol. 5(4), e1000352. [ Links ]
Ülger, I., Kamalak, A., Kaya, E. & Kurt, O., 2017. Comparison of chemical composition and anti-methanogenic potential of Liquidambar orientalis leaves with Laurus nobilis and Eucalyptus globulus leaves using in vitro gas production technique. Ciencia e Investigación Agraria 44(1), 75-82. [ Links ]
Ülger, I., Kaliber, M., Ayaçan, T. & Küçük, O., 2018. Chemical composition, organic matter digestibility and energy content of apple pomace silage and its combination with corn plant, sugar beet pulp and pumpkin pulp. S. Afr. J. Anim. Sci. 48(3), 497-503. [ Links ]
Xu, Q., Yuan, X., Gu, T., Li, Y., Dai, W., Shen, X., Song, Y., Zhang, Y., Zhao, W. & Chang, G., 2017. Comparative characterization of bacterial communities in geese fed all-grass or high-grain diets. PloS One 12, e0185590. [ Links ]
Zhao, L.L., Wang, G., Siegel, P., He, C., Wang, H.Z., Zhao, W.J., Zhai, Z.X., Tian, F.W., Zhao, J.X. & Zhang, H., 2013..Quantitative. Sci. Rept. 3, 1163. [ Links ]
Zhao, W., Wang, Y., Liu, S., Huang, J., Zhai, Z., He, C., Ding, J., Wang, J., Wang, H. & Fan, W., 2015. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PloS One 10, e0117441. [ Links ]
Zhou, H., Gao, Y., Gao, G. & Lou, Y., 2015. Oral administration of recombinant Lactococcus lactis expressing the cellulase gene increases digestibility of fiber in geese. Curr. Microbiol. 71(6), 693-698. [ Links ]
Zhou, H., Guo, W., Zhang, T. & Xu, B., 2018. Response of goose intestinal microflora to the source and level of dietary fiber. Poult. Sci. 97(6), 2086-2094. [ Links ]
Received 19 October 2019
Accepted 5 January 2020
First published online 13 July 2020
# Corresponding author: wangccq@foxmail.com


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}