










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Animal Science
versão On-line ISSN 2221-4062
versão impressa ISSN 0375-1589
S. Afr. j. anim. sci. vol.49 no.2 Pretoria 2019
http://dx.doi.org/10.4314/sajas.v49i2.7
ARTICLES
Genotype imputation as a cost-saving genomic strategy for South African Sanga cattle: A review
S.F. LashmarI, II, #; F.C. MuchadeyiII; C. VisserI
IDepartment of Animal and Wildlife Sciences, University of Pretoria, P/Bag X20, Hatfield, Pretoria, 0028
IIBiotechnology Platform, Onderstepoort Veterinary Institute, Agricultural Research Council, P/Bag X05, Onderstepoort, Pretoria, 0110
ABSTRACT
The South African beef cattle population is heterogeneous and consists of a variety of breeds, production systems and breeding goals. Indigenous cattle breeds are uniquely adapted to their native surroundings, necessitating conservation of these breeds as usable genetic resources to sustain efficient production of beef. Current projections indicate positive growth in human population size, with parallel growth in nutritional demand, in the midst of intensifying environmental conditions. Sanga cattle, therefore, are invaluable assets to the South African beef industry. Modern genomic methodologies allow for an extensive insight into the genome architecture of local breeds. The evolution of these methodologies has also provided opportunities to incorporate deoxyribonucleic acid (DNA) information into breed improvement programs in the form of genomic selection (GS). Certain challenges, such as the high cost of generating adequate numbers of dense genotypic profiles and the introduction of ascertainment bias when non-commercial breeds are genotyped with commercial single nucleotide polymorphism (SNP) panels, have caused a lag in progress on the genomics front in South Africa. Genotype imputation is a statistical method that infers unavailable or missing genotypic data based on shared haplotypes within a population using a population or breed representative reference sample. Genotypes are generated in silico, providing an animal with genotypic information for SNP markers that were not genotyped, based on predictive model-based algorithms. The validation of this method for indigenous breeds will enable the development of cost-effective low-density bead chips, allowing more animals to be genotyped, and imputation to high-density information. The improvement in SNP densities, at lower cost, will allow enhanced power in genome-wide association studies (GWAS) and genomic estimated breeding value (GEBV)-based selection for these breeds. To fully reap the benefits of this methodology, however, will require the setting up of accurate and reliable frameworks that are optimized for its application in Sanga breeds. This review paper aims, first, to identify the challenges that have been impeding genomic applications for Sanga cattle and second, to outline the advantages that a method such as genotype imputation might provide.
Keywords: breed improvement, developing countries, indigenous breeds, genomics
Introduction
South Africa accommodates a human population of approximately 57.7 million people (Statistics South Africa, 2018). According to UN projections, this number will increase to 72.8 million people by 2050 (United Nations, 2017). Nutritional demand, specifically the demand for animal protein, is expected to grow in parallel with population size and the responsibility of meeting this demand will weigh heavily on livestock production systems. Global warming will make bearing this responsibility a challenging task, with extreme environmental changes expected for developing countries south of the equator (Scholtz et al., 2013). Widespread regions of South Africa are experiencing a state of drought, which is the result of the strong El Nino event that occurred in the 2015/2016 year (South African Weather Service, 2016). The shortage of water in the country has raised concerns, among other things, about the amount of water it takes to finish cattle off in feedlots (Meissner et al., 2013). Approximately 68.6% of South African land is available for grazing, which is an ideal situation for extensive livestock production systems that rely on natural veld as feed source (Directorate: Knowledge and Information Management, 2017). Global warming, however, will be responsible for fluctuations in the nutritional value of the natural veld (Scholtz et al., 2014). Moreover, South Africa is geographically diverse and hosts a wide range of climatic zones, vegetation and soil types as well as a series of tick-borne and other endemic cattle diseases, each unique to its nine terrestrial biomes.
The South African beef population includes the Sanga cattle subspecies (Bos taurus africanus), which is indigenous. The breeds that belong to this subspecies include the Afrikaner, Drakensberger, Nguni and Tuli and the experimentally developed composite Bonsmara (Scholtz, 2010). These cattle, which are phenotypically distinguishable by their cervico-thoracic humps, resulted from historic crossbreeding between taurine and indicine cattle subspecies in eastern Africa (Payne & Hodges, 1997; Felius et al., 2014). From there, they were brought to southern Africa by migrating tribes (Schoeman, 1989), reaching South Africa by 250 - 500 AD (Payne & Hodges, 1997; Felius et al., 2014). Prolonged exposure to and endurance of the natural elements specific to South Africa have shaped the genomes of Sanga breeds to become habituated to the country's various extensive farming environments. Many trial-based studies have confirmed the ability of these breeds to adapt to, survive and reproduce in the varying beef-producing regions of South Africa. In the past decades several South African animal scientists have reported on and reviewed so-called proxy-indicator traits of adaptability for Sanga breeds. These studies have highlighted the potential of Sanga breeds to be highly fertile (e.g. Maule, 1973), calve easily and frequently (e.g. Scholtz, 1988; Schoeman, 1989; Collins-Lusweti, 2000), resist ticks and tick-borne diseases (e.g. Bonsma, 1980; Schoeman, 1989; Rechav & Kostrzewski, 1991), and be well suited to extensive finishing systems of South Africa (e.g. Du Plessis et al., 2006). The genetic architecture of SA Sanga breeds and the molecular mechanisms underlying their adaptation abilities, however, have been only partially investigated.
The number of genomics studies on beef cattle has grown steadily in South Africa over the past five years. The initial validation of the utility of the Illumina® bovine SNP50 bead chip for indigenous South African cattle by Qwabe et al. (2013) provided the first insight into genomics of Sanga cattle. Validation was important because no Sanga breeds were included in the SNP discovery process of this bead chip (Matukumalli et al., 2009). As was expected, lower minor allele frequency (MAF) was observed and consequently a lower number of informative SNPs for indigenous, non-discovery breeds and crossbreeds. Zwane et al. (2016) and Lashmar et al. (2018) confirmed the tendency towards low MAF in Sanga breeds using GeneSeek®'s GGP 80K and GGP 150K bead chips, respectively. In a series of successive studies, the authors who did the original validation, investigated the genetic diversity (Makina et al., 2014; Makina et al., 2016), linkage disequilibrium (LD) and effective population size (Ne) (Makina et al., 2015a) as well as selection signatures (Makina et al., 2015b) of indigenous breeds. The SNP50 bead chip was also utilized to identify copy number variations (CNVs) in Nguni cattle (Wang et al., 2015). Genes associated with biological processes such as immune and abiotic stress were among the genes identified within these CNV regions (Wang et al., 2015). These results were in partial agreement with the study by Makina et al. (2015b), in which it was revealed that a heat shock protein gene (HSPB9) and immune response genes were under selection in indigenous breeds. Mapholi et al. (2016) used SNP50 bead chip data to perform a GWAS to investigate tick resistance in a sample of close to 600 Nguni cattle. These authors identified several regions, in concordance with previous research, which harbour quantitative trait loci (QTL) that are linked to various tick-count traits. Only three of these regions, however, reached the threshold for genome-wide association significance, which necessitates further validation.
These studies provided a baseline for further investigation of SNP technology in South African beef cattle, especially for Sanga breeds. The local importance of these breeds, in terms of the role that they will play in enriching the South African beef industry, has attracted attention to the incorporation of genomic information into breed improvement programs in the form of GS. Genomic selection, however, relies on a good phenotypic and genotypic backbone. Owing to the relative size of the breed (about 120 000 animals) (Van der Westhuizen et al., 2014) and the completeness of phenotypic records, the SA Bonsmara was the first South African beef cattle breed for which GEBVs were estimated. This was preceded by a study that elucidated the population genetic structure within a possible reference population for the SA Bonsmara breed (Bosman et al., 2017). The Beefmaster, a synthetic breed composed of Brahman, Hereford and Shorthorn genetics (Porter, 1991), followed suit and was the second beef cattle breed for which GEBVs were released (Beefmaster Cattle Breeders Society of South Africa and SA Stud Book, 2017). The lack of accurate pedigree recording still prohibits the application of GS for a large portion of South African beef cattle breeds. Pedigree recording, however, has improved and for the Afrikaner, Drakensberger, Nguni and Tuli breeds the percentage of average pedigree completeness for first-generation animals is about 90% (Abin et al., 2016). Although performance testing has been available for commercial beef populations since 1959, and all indigenous breeds are participating actively, participation by some breeders' societies is as low as 32% (Scholtz, 2010; Van Marle-Köster et al., 2013).
Accumulation of sufficient genotypic information to initiate GS for breeds such as the SA Bonsmara was partly made possible through the Beef Genomics Project (BGP), which is a collaborative research-focused project that includes researchers from universities and research councils, breeders' societies and other role-players. The BGP is a large-scale project that is funded by the Technology Innovation Agency (TIA; Department of Science and Technology) with the overall aim of instating genomic improvement in the South African beef population (Becker, 2016). This funding initiative allowed the generation of approximately 7 000 genotypes across 16 breeds over a three-year period of routine genotyping (Van Marle-Köster & Visser, 2018). Before the BGP, the scale of genomic studies on beef cattle in South Africa was limited compared with international studies. South African studies have typically included small sample sizes and have focused on genome characterization and population genetic analyses. It is only since the inception of the BGP that improvements in the number of genotyped animals have made applications such as GS a possibility. The main reason for the lag in introducing these genomic technologies earlier for South African beef cattle was related to financial constraints. Even with major reductions in genotyping costs over the past decade, the per-animal price of genotyping is still expensive, especially for researchers in the developing world where adapted indigenous cattle resources, that often have unique and uncharacterized genomes, are located. Many local researchers therefore collaborate with international research groups to fund studies on indigenous breeds. Programs, such as BGP, therefore assists in mitigating the financial burden on individual research groups at local universities and research organizations to procure dense SNP genotypes towards realizing GS. Routinely generated genotypes could furthermore serve as a data resource for research groups with different research objectives that require large numbers of genotyped animals from certain breeds. These programs also assist in establishing intra-national collaborations and building local capacity.
Large numbers of genotyped animals are required to set up breed reference populations that are suitable for genomic evaluation. Post BGP, the financial responsibility of genotyping key animals in herds or populations will be that of interested breeders or farmers, and the current high costs of genotyping will probably impede further uptake of genomics if the benefits of this technology, in relation to the cost, cannot be realized. In contrast with the dairy industry (globally and in South Africa), where relatively few large breeds dominate the national herd, the beef industry is diverse and consists of approximately 30 breeds of taurine, indicine and Sanga descent, including taurindicine crosses and composite breeds (Strydom, 2008). These subspecies are diverse, with breeds belonging to each subspecies displaying distinct genetic and phenotypic characteristics. Breeds also differ in their population size and the traits recorded for, as breeding objectives are breeder society specific (Van Marle-Köster et al., 2013). Including DNA information in breed improvement is a key objective to achieve genetic gain for local cattle and to exploit the adaptive mechanisms they possess. The effective introduction of GS, however, has phenotypic and genotypic requirements. To fulfil the genotypic requirements would necessitate i) the generation of high-density genotypes to be more affordable; ii) the availability of more genomic profiles per breed (approximately 1000); iii) the ability to standardize between genotyping platforms; and iv) GS algorithms to be optimized for the genomic structure of local breeds. Genotype imputation is a methodology that uses predictive algorithms to enable the procurement of un-genotyped genomic information, and thereby allows for saving on genotyping costs. It is an ideal candidate methodology to assist in fulfilling the genotypic requirements for GS and, if properly validated and optimized for routine application, can accelerate genomics research in South Africa cost effectively.
This review aims to elucidate genotype imputation as a genomic strategy, focusing on its relevance to indigenous Sanga cattle breeds. The review discusses the methodology behind imputation, the factors that influence imputation accuracy, and its value in future applications such as GWAS and GS.
Genotype imputation
Genotype imputation is a statistical methodology that involves the prediction and simulation of missing SNP genotypes from observed or non-missing genotypes through model-based approaches (Marchini et al., 2007). The models are based on population genetic principles, and are used to deduce or extrapolate allelic correlations for sample sets with missing genotypic data using a sample set with dense and 'complete' data as a reference (Howie et al., 2009). Imputation of missing genotypes generally relies on the fact that extended haplotypes are shared over short distances between animals with a common ancestor (Pei et al., 2008). In effect, imputation therefore relies on family linkage, that is, the rules of Mendelian inheritance, or the LD structure within a population. If a reference population is genotyped on a high-density SNP panel and a test population is genotyped for a smaller subset of these SNPs, the assumption is that if they are related in some way, these populations should have similar underlying patterns of LD (Pei et al., 2008). The genotypic information of a reference population or sample is used to model patterns in genomic variation (Browning, 2008) and this genomic variation, which is typically shared within population, can therefore be used to infer missing genotypes in a non-reference animal or test sample.
Imputation therefore is population specific, and the accuracy with which genotypes can be imputed depends on the persistence of LD between animals in the reference and test populations. Imputation is hence viable only across breeds or populations if they are genetically similar or related in some way (Berry et al., 2014). Synthetic or composite breeds may therefore benefit from imputation if their component breeds are pooled in the reference population (Browning, 2008; Ventura et al., 2014). Imputing missing genotypes from a reference population that belongs to an ancestrally different breed from the test population will result in low-quality imputation and will negatively affect the reliability, and hence utility, of imputed genotypes (Browning, 2008).
If high-density genotypes can be imputed reliably from low-density SNP arrays with sufficient accuracy, this would allow for the opportunity to genotype more animals in a more affordable way (García-Ruiz et al., 2015). Imputation is therefore an important statistical tool for enriching applications such as GWAS and GS that require higher or more evenly distributed marker densities (Marchini et al., 2007). This tool will also enable comparison between SNP bead chip data that i) have been developed by different companies (e.g. AffymetrixTM, Illumina® and Neo-Geneseek®), and ii) that incorporate different SNP densities (e.g. 50K, 150K and 777K) in meta-analytic approaches (Ellinghaus et al., 2007). This can be done by identifying a set of SNPs that are common to two platforms and then imputing to a standard density. For example, if low- (e.g. 7K), medium- (e.g. 50K) and high-density (e.g. 150K) genotypic information is available for a breed, the low- and medium-density information can be imputed to the high density and this high-density information can be merged and collectively used for downstream analyses. This standardization might be useful when one considers the fast rate at which new and improved genotyping platforms are becoming available (Nicolazzi et al., 2015).
Imputation algorithms can generally be categorized as population-based and pedigree-based methods. Population-based algorithms assume large numbers of animals with unknown pedigree data and therefore rely on population-wide LD between markers (Weigel et al., 2010). These algorithms use a probabilistic approach to perform imputation and rely on shorter haplotypes that are typically less than 1 centiMorgan (cM) in length (Antolín et al., 2017). Probabilistic methodologies are ideal for natural populations and are more viable for high-density bead chips (Weigel et al., 2010; Mulder et al., 2012) because using methods that rely solely on LD information would be affected negatively when SNP densities are sparse and LD is low (Wang et al., 2016).
Pedigree-based algorithms use heuristic methods that assume the existence of family structure and rely on long haplotypes, typically larger than 10 cM in length, which are shared between closely related animals (Antolín et al., 2017). Imputation therefore cannot be reliably performed with these methods if accurate pedigree information is lacking. These methods are more suited to case-control studies or studies in which family trios - both parents and offspring - have been sampled. A number of imputation software programs exist, which differ in their approach to employing the methods discussed. The most popular imputation software programs in animal breeding and genetics research are listed in Table 1, which was adapted from Calus et al. (2014) and Antolín et al. (2017).
To summarize, probabilistic methods that rely on LD information generally use hidden Markov models (HMM) to perform imputation. HMMs are probabilistic models that allow assumptions and inferences to be made about hidden variables, each with a finite set of possible 'states', based on observable outputs (Rabiner, 1989). These models estimate the probability that a certain state could be responsible for producing a certain observable output at a given time (Rabiner, 1989). These models therefore use the underlying relationship, or correlation, between observed and unobserved SNPs to infer the most probable genotype for the unobserved SNPs. HMM methodology can be computationally demanding because it requires that genotypes are phased and estimate recombination rates. However, it can be supplemented with heuristic methodology that exploits more accurate phasing information owing to the incorporation of family linkage (Antolín et al., 2017). The addition of pedigree information, albeit not a necessity, will therefore boost imputation accuracy. In the same way, HMM methodology can add value to heuristic methods if pedigree information for only one parent is available.
The algorithms and exact methodology incorporated in each of the software programs (Table 1) are discussed in detail in each of the scientific papers and have been reviewed by for example Antolín et al. (2017) and Wang et al. (2016). Various statistical models have been proposed including HMM, haplotype clustering algorithms, linear regression models and expectation-maximization algorithms (Pei et al., 2008; Howie et al., 2009; Wang et al., 2016). Statistical methods differ in their approach to capturing haplotypes that are shared in a population (Pei et al., 2008). The choice of software therefore influences imputation accuracy. Several factors that are within and beyond the researcher's control might affect the quality of imputed genotypes.
Factors that affect imputation accuracy
The accuracy with which SNP genotypes can be imputed will determine the utility and reliability of a given imputation method for a given population. Parameters to quantify imputation accuracy can generally be subdivided into two groups, namely those that determine i) the proportion of alleles or genotypes that were correctly (e.g. Weigel et al., 2010) or incorrectly imputed (e.g. Druet et al., 2010; Zhang & Druet, 2010), and ii) the correlation or squared-correlation, usually the Pearson method of correlation, between true and imputed genotypes (e.g. Huang et al., 2009; Mulder et al., 2012; Ma et al., 2012). An alternative parameter, the imputation quality score (IQS), which determines imputation accuracy on the basis of statistics of agreements and adjusts for chance concordance has also been proposed (Lin et al., 2010). Methods that determine the concordance rate of imputed alleles (that is, proportion correctly imputed) will be more informative for imputing genotypes for GS, since GS algorithms assume additive allele effects (Berry et al., 2014). These methods, however, tend to inflate imputation accuracies because of sensitivity to low-frequency alleles (MAF<5%) (Ramnarine et al., 2015). Imputation of rare variants might therefore be better assessed using for example correlation-based accuracy measures that are less sensitive to MAF. All of these parameters are dependent on the availability of true and imputed genotypes, that is, scenarios in which a proportion of the true genotypes are available, but masked. A third category of accuracy quantification involves statistics that are built into programs such as BEAGLE (Browning & Browning, 2007), which do not require the availability of 'true' genotypic data, but rather estimate accuracies based on the likelihood or expectation of genotypes and allele dosage (Ramnarine et al., 2015).
Imputation accuracy depends on many factors including the imputation method, the MAF of the SNP to be imputed, LD between SNPs, the chromosomal position of the SNP (that is, whether it is located in the centre of the chromosome or on the chromosomal extremes), the quality of the SNP map, the discrepancy in SNP densities between high- and low-density SNP panels, and the size and composition of the reference population (Schrooten et al., 2014). Setting up a framework for incorporating imputation, as a routine genomic procedure, would require the optimization of imputation methodology for specific breeds or breed groups such as Sanga cattle. This might necessitate adjustments for example for national breed size (that is, numerically small versus numerically large) and variations that might occur across the genome, which might be the case for admixed populations. In addition, the algorithms that are built into certain imputation software might not be suited to the genomic architecture of certain breeds or populations. The main factors that need consideration are detailed below and are focused on the Sanga context.
Reference population size and degree of relatedness to the test population
The main consideration in the experimental design of an imputation study is determining an appropriate set of reference haplotypes or animals to achieve accurate predictions (Li et al., 2009). The most common method for assessing the influence of reference population size on the accuracy of genotype imputation is by masking varying subsets of reference animals and comparing the accuracies recovered for increasing reference population sizes (Li et al., 2009). Accuracy measures generally improve with larger reference population sizes, and this trend has been well documented (e.g. Hayes et al., 2012; Pausch et al., 2013; Piccoli et al., 2014; Ogawa et al., 2016). The improvement in accuracy, however, is not so pronounced for high-density bead chips compared with lower density ones (Ogawa et al., 2016). That is, the effect of reference population size is reduced if fewer genotypes are to be imputed. The improvement in imputation accuracies with increasing reference sample could be because more animals are used to construct haplotype libraries from which to impute. The inclusion of more reference animals increases the probability of including more breed or population representative haplotypes. The rule of thumb has generally been that a reference sample size of about 1000 animals will be sufficient for imputation. However, the ideal population size for a breed will depend on breed dynamics.
The breed dynamic of the South African beef population has seen significant changes in the past. In the 1970s, when the national herd was approximately 6 million head smaller than the approximately 13.6 million animals recorded today (Meissner et al., 2013), the indigenous Afrikaner dominated national herd numbers. Purebred Afrikaner and Afrikaner crosses represented approximately 70% of all the cattle slaughtered (Van Marle, 1974). Today, the experimentally developed SA Bonsmara composite is the most abundant breed in South Africa (about 120 000 registered animals) (SA Stud Book, 2016). The Nguni is the most abundant Sanga breed (about 38 000 registered animals) (SA Stud Book, 2016). Registered animals that belong to Sanga breeds such as the Afrikaner (about 7300 registered animals) (SA Stud Book, 2016), Drakensberger (about 12 800 registered animals) (SA Stud Book, 2016) and Tuli (about 9500 registered animals) (SA Stud Book, 2016) are far outnumbered by composite breeds such as the SA Bonsmara and SA Beefmaster (about 48 000 registered animals) (SA Stud Book, 2016).
The SA Bonsmara was the first South African beef cattle breed to receive genomic evaluations. This is attributable to the relative size and market share of the breed and superior record keeping by the breeders' society, and hence availability of phenotypes. In the BGP, the generation of genotypes per breed was conditional on the size and breeding objectives, the availability of phenotypic data, and the long-term prospects of implementing GS successfully (SA Stud Book, 2016). Approximately 42% of the animals participating in Logix Beef (SA Stud Book's animal recording database) belong to the SA Bonsmara, while only 2.4%, 4.5%, 5.6% and 3.3% of the animals that participate are Afrikaner, Drakensberger, Nguni and Tuli, respectively (SA Stud Book, 2016). The eventual aim is to implement GS for all these indigenous breeds. Establishing breed-appropriate reference populations, however, requires the accumulation of sufficient genotypic information as well. To date approximately 300, 1 850, 960, 400 and 200 genotypes have been generated for the Afrikaner, SA Bonsmara, Drakensberger, Nguni and Tuli breeds, respectively, during the timeframe of the BGP (personal communication). Genotypic profiles were mostly generated using the GeneSeek Genomic Profiler 150K bovine chip.
The ideal reference population is not influenced solely by the size of the reference population, but by its composition as well. Imputation accuracy improves if there is a level of relatedness between the reference and test samples. Berry & Kearney (2011) for example showed a positive correlation between the average relatedness between reference and test samples and imputation accuracy. This correlation strengthened when the maximum relatedness of test animals were considered instead of the average relatedness (Berry & Kearney, 2011). These authors observed an approximate 6% increase in genotype concordance rate for animals with both - as opposed to no - parents in the reference sample. Even though there are many imputation algorithms that can perform imputation fairly accurately for unrelated samples, the inclusion of parental or familial genotypes in the reference population will assist in boosting accuracy estimates. The reason for higher accuracy is due to closer linkage and therefore sharing of larger genomic segments within families. It would therefore be advisable or beneficial to include parent-offspring pairs or family trios in sampling efforts. This would be easier for dairy cattle, as opposed to beef, when one considers the higher prevalence of reproductive biotechnologies in the dairy industry.
In the SA Bonsmara for example strong genetic linkage between the animals that were genotyped was ensured by encouraging all breeders to submit hair samples of influential herd sires (SA Stud Book, 2017). Sampling was also done to ensure the inclusion of animals with accurate BLUP breeding values (SA Stud Book, 2017). This included female animals with superior breeding value accuracies for traits such as age at first calving, calving interval as well as maternal birth and weaning weights (SA Stud Book, 2017). Animals were selected across a spectrum of good and bad performers for these traits. The same process is utilized to select appropriate animals to genotype for other Sanga breeds. Although the focus of selection was not to specifically sample family trios, selecting genetically influential animals would indirectly assure the genotyping of animals that are directly related. If parent-offspring pairs (sire-offspring or dam-offspring) have been genotyped with BGP, additional funding could possibly be used to complete family trio genotypes by genotyping or imputing the missing parent. Berry et al. (2014) tested the imputation of parental genotypes based on their half-sib progeny and concluded that ungenotyped parental genotypes could be inferred accurately if genotypes were available for a sufficient number of offspring.
Genome resources and SNP arrays
There were originally two competing genome assemblies for cattle. The first reference genome, Btau_1.0, was assembled by the Human Genome Sequencing Centre at the Baylor College of Medicine (Elsik et al., 2009), whereas the Centre for Bioinformatics and Computational Biology at the University of Maryland initiated the assembly of an alternative genome, namely UMD2 (Zimin et al., 2009). Efforts to assemble these reference genomes occurred concurrently. The assembled genomes went through several stages of improvement over the years, resulting in the versions (Btau_5.0.1 and UMD3.1.1) that are currently available to researchers through the National Centre for Biotechnology Information (NCBI). At the 11th World Congress for Genetics Applied to Livestock Production the release of a new de novo assembly, ARS-UCD, of the Dominette Hereford genome was announced (Rosen et al., 2018). Long-read sequencing was utilized to reach 80X genome coverage with approximately 100- and 200-fold fewer gaps than the Btau_5.0.1 and UMD3.1 assemblies, respectively (Rosen et al., 2018).
From the two initial genome assemblies, SNP identification followed. Today various commercial SNP bead chips are available for cattle through three leading companies (Affymetrix™, Illumina®, Neogen's GeneSeek®) (Nicolazzi et al., 2015). The new assembly will aid in i) improving genome continuity, ii) remapping reads, and iii) improving marker ordering, which might influence SNP selection for the development of SNP genotyping platforms in the future (Rosen et al., 2018). The commercial bead chips that are currently available for cattle are summarized in Table 2, adapted from Nicolazzi et al., (2015). There are also a growing number of custom-made bead chips that are protected by intellectual property, and are therefore not available for commercial utilization (Nicolazzi et al., 2015).
Many of the bead chips that are listed in Table 2 have large numbers of SNPs in common. Some are updated versions of previously released bead chips, including additional SNPs that are optimized for specific purposes. Illumina's Bovine LD (6 909 SNPs), which was made available in 2011, for example has mostly replaced the Golden Gate 3K (2 900 SNPs), which was released in 2010 (Wiggans et al., 2013). A subtotal of 2 159 SNPs were retained from the Golden Gate (Wiggans et al., 2013). Geneseek recently released the bovine Genomic Profiler 150K (GGP 150K) SNP bead chip, which features 139 480 SNPs with an average inter-SNP distance of about 19 kilobase pairs (kb). The GGP 150K bead chip incorporates approximately 74 000, 42 000, 25 000 and 23 000 SNPs that are included on the original GGP HD (80K), Bovine SNP50 (Illumina), GGP LD and Bovine HD (777K, Illumina) bead chips, respectively. One of the most recently released platforms, the GeneSeek GGP Indicus bead chip, features about 35 000 indicine-specific SNPs that were selected from a cohort of breeds, including the Brahman, Nellore, Gyr and Santa Gertrudis and tropical composite breeds (Ferraz et al., 2018). The GGP indicus chip was also optimized for high imputation accuracy in indicine breeds, with imputation to the Illumina HD bead chip being up to 97% accurate in these breeds (Ferraz et al., 2018). Previous research has shown improved MAF and LD estimates for indigenous Ethiopian cattle (Edea et al., 2015) and improved imputation accuracy for indicine Gyr cattle (Boison et al., 2015) when indicus-derived SNP panels were used. Since Sanga cattle are taurine-indicine hybrids, it would be recommended to test the utility of this panel for Sanga breeds, in contrast to taurine-derived SNP panels. Lower-density bead chips in general are increasingly being developed with the aim of retaining specific subsets of SNPs, in common with higher density bead chips, to be optimized for low-cost genomic applications (Boichard et al., 2012).
The bovine reference genome will undergo many updates in the future as sequencing technologies improve. This is an important consideration because re-mapping of SNPs can cause rearrangements in the haplotypes captured by bead chips, especially if SNPs are re-mapped to different chromosomes (Milanesi et al., 2015). Incorrect SNP positioning of more than 5 000 SNPs on the bovine HD bead chip has been suggested (Pausch et al., 2013). Pre-imputation, ensuring consistency between SNP positions of low- and high-density panel genotypic data is an important quality check. Software such as the web-based tool SNPchiMp (Nicolazzi et al., 2015) provides a platform for standardizing SNP genomic positions by mapping markers to the same reference genome (that is, either to the UMD3.1, Btau_5.0.1 or ARS-UCD genome assemblies). Accurate mapping of SNPs is also important owing to the decrease in imputation accuracy, which has been observed for SNP genotypes on chromosomal extremes, as opposed to SNPs that are located in the centre of the chromosome, in previous studies (e.g. Ventura et al., 2016).
Studies that focus on Sanga beef cattle have utilized Illumina SNP50 (e.g. Qwabe et al., 2013), GeneSeek GGP 80K (e.g. Zwane et al., 2016) and GeneSeek GGP 150K (e.g. Lashmar et al., 2018) genotypic data for various genomic applications. Since the initiation of the BGP, data has been routinely generated using the GeneSeek GGP 150K panel, whilst specific research projects have generated low-density (e.g. Illumina 7K) and high-density (e.g. Illumina HD) genotypic data and whole-genome sequencing information, depending on the research interest. The diversity in genomic data that is available for Sanga cattle might therefore require standardization between platforms in future efforts to combine data in meta-analyses and collaborative projects.
The discrepancy in the number of SNPs between panels also influences imputation accuracy, specifically between low- and high-density panels (that is, the number of SNPs to be imputed). Accuracy estimates tend to improve with increasing SNP density of the low-density panel. That is, the fewer the number of SNPs that need to be imputed, the higher the mean imputation accuracy will be. This increase in accuracy can be attributed to the fact that haplotypes can be more accurately resolved with more SNPs present (Tsai et al., 2017). Imputing Dutch Holstein genotypes to a custom 60K bead chip, Zhang & Druet (2010) observed decreasing imputation error rate when the SNP density of the low-density panel was improved from 384 SNPs to 6 000 SNPs. These authors then suggested a minimum of 3 000 SNPs to achieve 3% or lower imputation error rate (Zhang & Druet, 2010). Ogawa et al. (2016) indicated a similar trend in imputing to 50K genotypes for Japanese Black beef cattle. Imputation accuracy was 2.7% higher when the low-density panel included 10 000 SNPs versus 500. This relationship agrees with imputation experiments on sheep (Hayes et al., 2012), salmon (Tsai et al., 2017) and maize (Hickey et al., 2012), and suggests that a minimum set of SNPs is required to allow optimal imputation.
The minimum number of SNPs that are necessary for accurate imputation must be determined to develop an imputation-driven low-density panel for Sanga cattle. For individual breeds, this will depend on the extent of LD within the population. Breeds that are characterized by lower LD will require higher SNP densities. Developing a low-density panel that is applicable across Sanga breeds will also depend on the persistence of LD across Sanga breeds. If the persistence of LD across breeds is low, a higher number of SNPs would be necessary as a minimum for the low-density panel in imputation.
Pre-imputation processing of genotypes
Pre-imputation procedures such as quality control (QC), DNA strand checking and phasing aid in processing and preparing genotypic data to optimize imputation accuracy. QC is an important first step in any genomics study, and serves to remove uninformative samples and markers in preparation of downstream analyses. Sample-based QC will exclude individual animals with discordant sex information (when pedigree- versus SNP-based gender assignment disagree), that have high percentages of missing genotypes (have a low call rate), display outlying heterozygosity rates and show evidence of non-Mendelian inheritance (Li et al., 2009; Anderson et al., 2010). Marker-based QC involves excluding SNPs with low genotype call rate, those that deviate significantly from Hardy-Weinberg equilibrium, those with low MAF, and those that have duplicated or unknown genomic positions (Anderson et al., 2010; Purfield et al., 2016). The stringency of the quality filters applied prior to imputation may influence the accuracy of imputed SNPs. Roshyara et al. (2014) proposed that for small to moderate datasets, less stringent or no QC procedures prior to imputation would be best practice owing to the detrimental effect that stringent QC procedures might have on the quality of imputation for such datasets. More stringent QC procedures might also discard SNPs that could have been successfully imputed (Roshyara et al., 2014) or rare SNPs that are informative for the expression of traits of interest, such as those pertaining to the adaptive mechanisms of Sanga cattle. Purfield et al. (2016) for example investigated the effect of sample call rate as a QC parameter on imputation accuracy, and observed improved genotype and allele concordance rates with increasing animal call rate. Genotype concordance rates improved from 0.41 to 0.95 when animal call rates were <40% versus when call rates were between 95% and 99% (Purfield et al., 2016). These authors consequently proposed a cut-off of 85% as the lower limit for exclusion of animals based on call rate. QC procedures should therefore be optimized to retain high-quality data, but should not compromise the representation of SNPs in haplotype libraries used for imputation.
The quality of genotype imputation depends on whether the allele calls that are being generated for the test population are from the same physical DNA strand in relation to the reference genome (Verma et al., 2014). Determining the DNA strand orientation is therefore an essential pre-processing step. SNP annotations also differ for datasets, and on the genotype platform and the genotype-calling algorithm (Verma et al., 2014). Illumina for example uses a TOP/BOT method, which designates top (TOP) and bottom (BOT) DNA strands based on the SNP and its flanking sequence, and calls alleles based on a generalized 'Allele A' and 'Allele B' nomenclature (Illumina, 2006). Genotypic information can also be provided in a forward/reverse orientation. Inconsistencies between genotypic data between the reference and test populations with regard to strand orientation and allele coding - that is, whether genotypes are coded as A/B or A/C/G/T format - can impede accurate imputation. Certain imputation software programs, such as BEAGLE (Browning & Browning, 2007), IMPUTE2 (Howie et al., 2009), and PLINK (Purcell et al., 2007), have DNA strand checking utilities to determine strand orientation and to subsequently convert to a flipped strand orientation when necessary. During this procedure, alleles are converted to their complements based on observed alleles and the MAF and LD pattern that is observed for SNPs, and then removed when inconsistencies in these parameters cannot be resolved (Verma et al., 2014). The developers of SNPchiMp (Nicolazzi et al., 2015) have made bioinformatics tools available in the form of an application called SNPConvert, which can also assist researchers in standardizing allele coding and strand orientations for SNP genotypic data.
Haplotype phasing involves determining from which of the parental chromosomes or haplotypes SNP alleles are derived or on which they are located (Browning & Browning, 2011). SNP genotypic data is generally unphased and for the purpose of imputation it is essential to know the origin and location, i.e. on which DNA strand, of each allele of a bi-allelic SNP (Browning & Browning, 2011). Accounting for unknown phase is, however, computationally intensive and can be time-consuming (Howie et al., 2012). Some imputation software such as BEAGLE (Browning & Browning, 2007), however, performs phasing as part of the imputation process. In other cases, third-party software such as SHAPEIT (O'Connell et al., 2014) is available for 'pre-phasing' genotypic data in a two-step imputation approach. In the two-step approach, observed genotypes are firstly phased and then the phased genotypic information is used for imputation. Pre-phasing will be useful in speeding up computation time of the overall imputation process but the value thereof will depend on the accuracy with which haplotypes can be estimated (Howie et al., 2012). Nevertheless, haplotype phasing will become increasingly important in future efforts to impute to sequencing data and the methods currently available are comprehensively reviewed in Browning & Browning (2011).
Population-specific parameters: minor allele frequency
Minor allele frequency (MAF) can have significant effects on the reliability of imputation, and a number of authors have investigated the influence of varying levels of MAF on imputation accuracy (e.g. Hozé et al., 2013; Schrooten et al., 2014; Van Binsbergen et al., 2014). The effect of low MAF versus high MAF on imputation accuracy will be determined primarily by how 'accuracy' is defined, that is, whether it is quantified as a proportion of correctly imputed genotypes or as a correlation between observed and imputed genotypes. It has consistently been found that accuracy quantified as a proportion or percentage of correctly imputed genotypes is correlated negatively with increasing MAF, whereas the correlation-based measure shows a positive relationship. Investigating different maize lines, Hickey et al. (2012) observed decreasing percentage-based accuracy, as opposed to increasing correlation-based accuracy, for increasing levels of MAF. For proportion-based or percentage-based measures, it is debated that when the frequency of the minor allele is low, there is a greater likelihood that imputation algorithms will predict genotypes as homozygous for the major or common allele (Hickey et al., 2012). Conversely, high MAF creates more uncertainty, thereby deeming predictions less reliable and hence less accurate. The correlation-based measure is less dependent on allele frequency and assumes that low-MAF SNPs are not sufficiently segregating within the population, and therefore cannot be easily imputed based on shared haplotypes (Hickey et al., 2012). Similar relationships were observed in European cattle, where Ma et al. (2012) indicated a higher 'correct rate' for lower MAF versus lower correlations for lower MAF across six widely used imputation software programs. These authors also observed that software that incorporated pedigree information was more sensitive to variation in MAF (Ma et al., 2012). In pigs, Badke et al. (2013) indicated the same trends with regard to the relationship between accuracy measures and MAF, but found that proportion-based measures also improved with increasing MAF when inter-SNP differences in MAF are adjusted for.
Factors pertaining to the experimental design of imputation studies may influence the effect size of MAF on imputation accuracy. These include the size of the reference population and the method of imputation implemented within software programs. Imputation accuracy can be improved, and error rate lowered (Huang et al., 2009), for rare SNP if a larger, more extensive reference population is used (Howie et al., 2009). Allele frequencies of rare SNP are typically overestimated when a small reference population is used (Howie et al., 2009). Possible adverse effects that low MAF might have on imputation accuracy will therefore be gradually alleviated with increasing animal numbers and improved representation.
MAF is essentially an indication of whether a SNP is segregating within a given population and therefore the composition of the reference population plays an important role as well. Boichard et al. (2012) indicated higher MAF, and therefore higher imputation accuracy, for cattle breeds that were used to design the bead chip under study. In humans, Howie et al. (2011) observed that although population-specific reference panels tend to outperform HapMap panels for imputation accuracy, reference panels that are 'ancestrally inclusive' and non-specific, may capture poorly represented low-frequency alleles. This would be important when genotypes need to be imputed for composite or crossbreeds of uncertain or unknown genetic composition such as the Drakensberger Sanga breed. Alleles that occur in low frequencies are not necessarily presented in reference haplotypes and therefore certain imputation software may have difficulty deriving the correct allele, which would affect the reliability of accuracy estimates directly (Schrooten et al., 2014). Software programs differ in their ability to detect copies of the minor allele. Howie et al. (2009) showed that some methods are more prone to erroneous minor allele calls. Certain software programs are better equipped to deal with low-frequency SNPs. The use of the appropriate method to optimize imputation for non-commercial breeds, which might be disadvantaged by ascertainment bias, would be an important consideration.
Lower average MAF has been observed for Sanga versus exotic breeds, verifying the existence of ascertainment bias (Qwabe et al., 2013). Furthermore, higher MAF was observed for a Sanga crossbreed (Angus x Nguni) than for a 'pure' Sanga breed (Nguni) (Qwabe et al., 2013). The use of a commercial bead chip such as the Illumina bovine SNP50 might therefore be more useful for crossbreeds that carry taurine haplotypes and therefore display higher MAF for the SNPs that were discovered for the chip. In imputation for instance this will be useful only when these haplotypes are represented by an appropriate reference sample that is, either of the component breeds or high-impact animals from the crossbred population. Investigating the impact of MAF on imputation accuracy is important not only because of its direct impact, but also because of the influence it may have on other factors such as LD that determine imputation accuracy. Incorporating imputation as a genomic strategy will therefore require a complete understanding of the complex interplay between various population-specific parameters. Low-MAF SNPs have been observed to underestimate r-squared (r2) based LD estimates (e.g. Khatkar et al., 2008; Qanbari et al., 2010), and LD has been shown to increase with increasing MAF for Sanga breeds (e.g. Makina et al., 2015a; Lashmar et al., 2018). In addition to parameters that characterize individual SNPs, it is important to look at the genomic relationship between SNPs.
Population-specific parameters: linkage disequilibrium and effective population size
Among the factors that have an influence of imputation accuracy, LD is probably the most important and has the potential to be limiting to achievable accuracy. The importance of LD, as a determining factor of imputation accuracy, has previously been shown where the influence of MAF was diminished in regions of high LD (Pei et al., 2008). The ability to impute a given genotype is affected directly by the strength of local LD in the genomic region in which that SNP is located (Hickey et al., 2012). Consensus has been that stronger LD improves imputation accuracy. Imputation algorithms are able to more accurately identify the haplotypes that are present on each gamete for individuals that are genotyped with a low-density panel, when LD is high (Hickey et al., 2012). Low inter-SNP LD is generally characteristic of populations with large effective population sizes (Bovine HapMap Consortium, 2009). This has been shown to impede accurate imputation (Pausch et al., 2013). If LD is weak and does not persist over long genomic segments, finding key ancestors that are representative of the breed becomes difficult. Weak persistence of phase and LD across breeds also limits the application of multi-breed imputation. For populations that display weak LD, however, algorithms have been proposed to simulate LD specifically for association studies (Yuan et al., 2011). This presents an opportunity to test imputation accuracy on different levels of LD.
Makina et al. (2015a) revealed LD of r2>0.2 to extend to an inter-SNP distance of 100 kb in the Afrikaner breed, while the same level of LD extended only to a distance range of 10 - 20 kb for other Sanga breeds such as the Drakensberger and Nguni. This corresponds to the relative sizes of these breeds in the national beef herd of South Africa - the Afrikaner is numerically the smallest of the Sanga breeds. Given a standard r2value of 0.2, which has been proposed as the ideal r2for GS and association studies, the Drakensberger and Nguni breeds would require approximately 150 000 SNPs, as opposed to 30 000 SNPs for the Afrikaner, for within-breed analysis (Makina et al., 2015a). The utility of the 150K GGP bovine bead chip therefore needs consideration for the Drakensberger and Nguni breeds.
The Ne of a population gives an indication of the evolution of a breed and can assist in understanding the genetic architecture that underlies traits (Falconer & Mackay, 1996). This parameter essentially gives an indication of the number of animals within a breed that contribute to the genetic makeup of the national herd. The Ne is dependent on the interplay between LD and the recombination distance between SNPs, in which the LD across a greater distance will be indicative of more recent Ne and LD across a shorter distance will indicate Ne in the more distant past or ancestral Ne (Barbato et al., 2015). SNPs would be more accurately imputed for breeds with smaller Ne, which display higher within-population LD and therefore share larger haplotypes.
Makina et al. (2015a) observed Ne estimates of 41, 87 and 95 for Afrikaner, Drakensberger and Nguni breeds, respectively. These estimates were higher than those observed for exotic breeds such as Angus and Holstein. The discrepancy can be expected, because exotic beef breeds were generally subjected to intense artificial selection much earlier, and more consistently, than local commercial breeds. In comparison with dairy breeds, beef breeds are extensively managed, and breeding practices rely considerably less on reproductive technologies such as AI and MOET, and generally rely on natural mating. According to SA Stud Book's 2016 annual report, 31% of the SA Angus births resulted from AI, while only 0.5%, 8% and 1.6% of Afrikaner, Drakensberger and Nguni calves were born from this technology (SA Stud Book, 2016). The variation in Ne estimates between Sanga breeds can be explained by the higher level of admixture observed within Drakensberger and Nguni breeds in comparison with the Afrikaner breed (Makina et al., 2014). In admixed genomes, a higher number of small haplotypes will be shared, as opposed to a smaller number of long genomic segments. The Afrikaner breed has experienced a significant decline in its population size over the past decades. This is postulated to be a result of increased utilization of the breed to develop composites, causing a small number of 'pure' Afrikaner animals to remain (Pienaar et al., 2014). It is important to consider Ne within the context of the actual or census population size within the national herd. The Afrikaner breed went from being the most abundant indigenous breed in the 1970s (Pienaar et al., 2014) to the numerically smallest Sanga breed, consisting of only 42 herds and approximately 7 300 animals nationwide (SA Stud Book, 2016). The SA Bonsmara for example is currently the most numerous indigenous breed in South Africa with numbers of upwards of 120 000 animals. However, it has an estimated Ne of 77 (Makina et al., 2015a). On the contrary, the national Drakensberger herd is approximately a tenth of the size, with an estimated Ne of 87. In the national herd of South Africa, it might therefore in theory be easier to sample animals that contribute to the population-wide genomic profile of the Drakensberger breed, and thereby achieve higher imputation accuracies, if high-impact animals can be identified and records are complete and accurate.
The utility of imputed SNPs in improving genomic applications
Genome-wide association studies
Genome-wide association studies (GWAS) aim to locate QTL or genes responsible for traits of economic interest. These studies use association signals between phenotypic and genotypic (genome-wide SNPs) information to guide researchers towards the location(s) on the genome responsible for expressing traits of interest (Hayes & Goddard, 2010). These candidate regions can then be used as a reference to search for causative SNPs that explain a proportion of the variation that is seen in that trait (Hayes & Goddard, 2010). This methodology may have significant implications for cattle, because it can be used to better understand the genetic mechanisms underlying economically important traits, such as those involved in the adaptability of Sanga cattle. It can also be a diagnostic tool to identify SNPs that are associated with cattle disorders, which can then be used to select against animals that carry deleterious alleles. The utility of SNPs that are identified by GWAS, however, needs to be verified in an independent set of animals to determine their validity and reproducibility. After verification, these SNPs may then be included in commercial or population-specific and production-specific SNP panels. These SNPs, however, may still not be informative for the entire spectrum of cattle breeds if they are monomorphic and both alleles are not segregating within a population.
It has been proposed that most traits follow a trend of 'common disease-common SNP'. However, common SNPs have been shown to have limited influence on complex diseases in humans (Pritchard & Cox, 2002). The association signals that are observed for common SNPs may be synthetic, meaning that these signals might not be influenced by common SNPs, but rather by rare SNPs that are in strong LD with common SNPs (Pritchard & Cox, 2002). Rare SNPs could be causative variants, but might not be - and in most cases are not - included on the bead chips that are commonly used to perform GWAS. The association of rare SNPs with common diseases or phenotypes is difficult to capture owing to poor statistical power, and generally requires genotyping of large numbers of animals. The alternative is to capture these SNPs directly by using higher density SNP panels or through whole-genome sequencing efforts. Re-sequencing of whole genomes, which is aimed at discovering novel SNPs, is not always feasible because of the requirement for large datasets of sequenced animals and the relative cost of sequencing per animal (Van Binsbergen et al., 2014). Methods such as genotyping by sequencing (GBS), which uses restriction enzymes to target specific segments of the genome, have been proposed to reduce sequencing costs and complexity. Genotyping by sequencing, however, has the limitation of producing high volumes of missing data, owing to the presence of variation in restriction sites, resulting from factors such as genetic divergence and low sequence coverage (Brouard et al., 2017). This presents the opportunity for the utilization of methods such as imputation to fill the missing data gaps.
The main purpose of imputation for GWAS is to boost the number of SNPs that can be tested for association and hence improve the power of the study (Marchini & Howie, 2010). Because animals can be genotyped on lower density panels, which can then be imputed to SNP densities that equated to a high-density panel or GBS and sequencing data, more animals can be affordably genotyped and included for analyses. Imputation has proved to increase power by up to 10% for GWAS, with rare SNPs being proposed to gain the most from this method (Marchini et al., 2007; Spencer et al., 2009; Marchini & Howie, 2010). Furthermore, after signals of association have identified certain genomic regions of interest, imputation can assist in fine mapping these regions, which will improve the chances of identifying the causal SNP or SNPs directly (Marchini & Howie, 2010). In the past, in many cases GWAS results were not replicable because the cost of SNP genotyping limited sample size, which limits comparability, and also owing to the availability of SNP data from different panels. Imputation, however, can be utilized to standardize the number of SNPs from different studies by imputing to a common set of SNPs to allow meta-analysis at each given SNP locus (Marchini & Howie, 2010). The combination of multiple datasets aims to reduce the number of false positive associations (Begum et al., 2012). Meta-analysis has been applied successfully, and has resulted in the identification of new loci of interest that had not been identified previously in individual studies (Marchini & Howie, 2010). These studies have been limited for cattle with only few meta-analyses performed for beef (e.g. Minozzi et al., 2012; Bolormaa et al., 2014). All of these studies, however, have identified novel genomic regions of interest when using merged data sets. The only way to truly capture novel regions of interest or novel SNPs, however, is through genome re-sequencing.
A number of studies have been published that investigated the utility of imputed sequence variants for cattle (e.g. Van Binsbergen et al., 2014; Frischknecht et al., 2017; Pausch et al., 2017; Bernardes et al., 2018). Imputation to sequencing data has been simplified by the initiation of the 1 000 Bull Genomes Project in 2012, which provides a database of whole-genome sequence information that is made available to research groups that are interested in imputation towards GWAS and GS (Daetwyler et al., 2014). Sequencing variants have largely been imputed from Illumina's Bovine HD SNP panel. Imputation accuracies are compromised when imputing from lower density SNP panels such as the SNP50 panel, and hence a two-step procedure has been proposed, imputing first from SNP50 to HD, and subsequently from imputed HD to sequencing data (Bernardes et al., 2018). Imputation to GBS data, which achieved up to 94% accuracy estimates, has also been tested in for example Canadian dairy cattle (Brouard et al., 2017). Re-sequencing data that was generated for Sanga breeds from studies such as Zwane (2017), which aimed to identify novel SNPs, provides a valuable resource for future South African studies that aim to utilize imputed sequence variants in GWAS and GS experiments.
Cost-effective genomic selection
Genomic selection, a concept that was first proposed by Meuwissen et al. (2001), is a method that incorporates dense SNP genotypes to estimate GEBVs (Hayes et al., 2009). A reference population with known SNP information and adequate performance and pedigree data is used to compile a prediction equation for the estimation of GEBVs in selection candidates (Meuwissen et al., 2001). GS essentially captures all locus effects, regardless of size, that contribute to the genetic variation for a trait of interest by summing all estimated effects across the entire genome into a GEBV (Hayes et al., 2009). These GEBVs are then used to aid selection decisions. The various models whereby GEBVs are estimated have been discussed in detail in previous literature (e.g. Goddard & Hayes, 2007; Goddard et al., 2010; Van Marle-Köster et al., 2013).
The four main driving factors that influence GEBV accuracy for a population are i) the population-wide LD; ii) the availability and completeness of genotypic and phenotypic data for the reference population; iii) the heritability of the trait in question; and iv) the distribution of QTL effects (Hayes et al., 2009). The latter two factors are subject to the trait being studied. The decay of LD with increasing inter-marker distance has been reported widely, and specifically for non-exotic and admixed populations (e.g. Lashmar et al., 2018; Edea et al., 2015; Mokry et al., 2014). Owing to the costs of acquiring high-density SNP genotypes and whole-genome sequencing data, especially for researchers in developing countries, improving SNP densities through genotyping and sequencing more SNPs is not economically feasible. Genotype imputation, however, will aid in cost-effectively improving SNP densities by genotyping animals with low-density panels and imputing to higher-density information. This would enable more efficient use of funds and genotyping more animals. Cost efficient genotyping will aid in the procurement of the rule of thumb 1 000 animals required for accurate GS (Meuwissen et al., 2001) and thereby availing this technology for possibly all Sanga breeds. The availability of sufficient performance and pedigree records, however, might then become the limiting factor. For this reason, the implementation of GS was first focused on Sanga breeds with good histories of animal recording such as the SA Bonsmara.
Developing a working pipeline of cost-effective GS for other Sanga breeds will require a low-density panel, consisting of Sanga-informative SNPs, which is optimized for accurate imputation. SNPs to be included on such a low-density panel will need to be selected from a pool of SNPs that are already included on high-density panels or have been sequenced for Sanga breeds, based on certain marker characteristics. These characteristics include the genome distribution (that is, whether SNPs are evenly spaced across the genome), the MAF (that is, whether SNPs are segregating within the population) and the LD pattern between SNPs. Methods that combine these attributes, such as the Wellman SNP selection method (Wellman et al., 2013), and methods that incorporate machine-learning algorithms, such as feature similarity (Phuong et al., 2006), have been used to select informative SNPs for Irish cattle (judge et al., 2016). Wu et al. (2016) also developed a multi-objective local optimization (MOLO) method for SNP selection, which uses a function that adjusts for gaps in the genomic data and incorporates Shannon entropy and other attributes, such as MAF and distribution, to select optimal SNPs. These methods need to be tested and validated for Sanga cattle to identify the optimal way of selecting Sanga-informative SNPs. Once the optimal SNP selection method and the optimal density (i.e. the minimum number of SNPs necessary from which to impute) have been identified, a low-density panel can be developed. This panel would serve as a backbone for using imputed SNPs in GS, which would allow the estimation of GEBVs at a reduced cost.
The utility of imputed genotypes for GEBV estimation has been studied for beef cattle (e.g. Berry & Kearney, 2011; Cleveland et al., 2011; Mulder et al., 2012). Mulder et al. (2012) confirmed the feasibility of direct genomic value (DGV) estimation using low density bead chips, provided that these bead chips included at least 3 000 SNPs. Cleveland et al. (2011) observed some loss in the accuracy of GEBVs using imputed SNPs, but still retrieved accuracy estimates that were higher than those acquired by traditional BLUP. Berry & Kearney (2011) observed a correlation of 97% between DGVs estimated from true genotypes versus imputed ones across a set of 15 functional traits. The correlations, however, depend on the availability of records for a specific trait and hence the reference population size that is used to estimate the SNP effects for that trait (Berry & Kearney, 2011). If more records are available, which is usually the case for easy-to-measure traits (e.g. weaning weight), then more animals can be included in the reference sample and the higher the DGV correlation from true genotypes versus imputed ones. For certain traits (e.g. direct and maternal calving difficulty), low DGV correlations are observed, regardless of relatively large reference population size. This phenomenon may be attributed to large QTLs being responsible for the expression of these traits (Berry & Kearney, 2011). Rutkoski et al. (2013) observed that the most accurate method of imputation was not necessarily always responsible for the most accurate GEBV estimation. This was ascribed to non-random imputations errors. These errors can be indicative of possible genetic relationships in the GS model if they are shared between related animals (Weigel et al., 2010; Rutkoski et al., 2013). Small numbers of wrongly imputed SNPs, however, are expected to have a negligible effect on GEBV accuracy since GS estimates all SNP effects simultaneously, as opposed to the SNP-by-SNPs approach that is followed in GWAS (Badke et al., 2013). Nevertheless, imputation as a methodology needs to be optimized to minimize, as far as possible, the effect of imputation variability on GS endeavours, especially for breeds with heterogeneous genomes such as those that belong to the Sanga subspecies.
Conclusion
The promotion of locally adapted Sanga breeds relies on the utilization of genomics in breed characterization and improvement. Imputation provides a cost-saving strategy for applying genomic methodologies such as GWAS and GS that will aid in breed improvement. Implementation of imputation as a routine genomic strategy, however, relies on its accuracy and hence reliability, which is influenced by many variables. The incorporation of imputation would therefore require optimization in Sanga breeds. Once a working pipeline has been set up for utilization, this methodology would hold many advantages for downstream genomic applications that aim to advance indigenous South African beef breeds.
Acknowledgements
The research project that encompasses this review is funded by the Red Meat Research & Development (RMRD-SA) and the Beef Genomics Project (BGP). The financial assistance of the NRF towards this research is also acknowledged. Opinions expressed and conclusions arrived at are those of the authors, and are not necessarily to be attributed to the NRF.
Author's Contributions
SFL formulated and refined the review article as part of his PhD: Animal Science project. CV and FCM supervised SFL and were responsible for revising, editing and structuring the article.
Conflict of Interest Declaration
None of the authors has a conflict of interest to declare.
References
Abin, S.A., Theron, H.E. & Van Marle-Köster, E., 2016. Population structure and genetic trends for indigenous African beef cattle breeds in South Africa. S. Afr. J. Anim. Sci. 46, 152-156. [ Links ]
Anderson, C.A., Pettersson, F.H., Clarke, G.M., Cardon, L.R., Morris, A.P. & Zondervan, K.T., 2010. Data quality control in genetic case-control association studies. Nat. Protoc. 5, 1564-1573. [ Links ]
Antolín, R., Nettelblad, C., Gorjanc, G., Money, D. & Hickey, J.M., 2017. A hybrid method for the imputation of genomic data in livestock populations. Genet. Sel. Evol. 49, 30. [ Links ]
Badke, Y.M., Bates, R.O., Ernst, C.W., Schwab, C., Fix, J., Van Tassell, C.P. & Steibel, J.P., 2013. Methods of tagSNP selection and other variables affecting imputation accuracy in swine. BMC Genet. 14, 8. [ Links ]
Barbato, M., Orozco-terWengel, P., Tapio, M. & Bruford, M.W., 2015. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet 6, 109. [ Links ]
Becker, J., 2016. Beef genomics programme: sequencing for bovine superiority. Available online at URL: http://www.livestockgenomics.co.za. [ Links ]
Beefmaster Breeders' Society of SA & SA Stud Book, 2017. Joint media release: Genomic breeding values for South African Beefmaster cattle. Available online at: http://www.sastudbook.co.za/images/photos/News-Beefmaster-Genomic-EBVs.pdf [ Links ]
Begum, F., Ghosh, D., Tseng, G.C. & Feingold, E., 2012. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. 40, 3777-3784. [ Links ]
Bernardes, P.A., Al-Mamun, H.A., Suarez, M., Lim, D., Park, B. & Gondro, C., 2018. Imputation accuracy of whole-genome sequence data in Hanwoo cattle. In: Proceedings of the 11th World Congress of Genetics Applied to Livestock Production. Auckland, New Zealand, 6-11 February 2018. [ Links ]
Berry, D.P. & Kearney, J.F., 2011. Imputation of genotypes from low-to high-density genotyping platforms and implications for genomic selection. Animal 5, 1162-1169. [ Links ]
Berry, D.P., McClure, M.C. & Mullen, M.P., 2014. Within- and across-breed imputation of high-density genotypes in dairy and beef cattle from medium- and low-density genotypes. J. Anim. Breed. Genet. 131, 165-172. [ Links ]
Boichard, D., Chung, H., Dassonneville, R., David, X., Eggen, A. & Van Tassell, C.P., 2012. Design of a bovine low- density SNP array optimized for imputation. PloS one 7, e34130. [ Links ]
Boison, S.A., Santos, D.J.A., Utsunomiya, A.H.T., Carvalheiro, R., Neves, H.H.R., Perez O'Brien, A.M., Garcia, J.F., Sölkner, J. & da Silva, M.V.G.B., 2015. Strategies for single nucleotide polymorphism (SNP) genotyping to enhance genotype imputation in Gyr (Bos indicus) dairy cattle: Comparison of commercially available SNP chips. J. Dairy Sci. 98, 4969-4989. [ Links ]
Bolormaa, S., Pryce, J.E., Reverter, A., Zhang, Y., Barendse, W., Kemper, K., Tier, B., Savin, K., Hayes, B.J. & Goddard, M.E., 2014. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 10, e1004198. [ Links ]
Bonsma, J.C., 1980. Cross-breeding, breed creation and the genesis of the Bonsmara. Livestock production. A Global Approach. Ed. J.C. Bonsma. Tafelberg, Cape Town, South Africa. pp. 126-136. [ Links ]
Bosman, L., Van Marle-Köster, E., Van der Westhuizen, R.R., Visser, C. & Berry, D.P., 2017. Population structure of the South African Bonsmara beef breed using high density single nucleotide polymorphism genotypes. Livest. Sci. 197, 102-105. [ Links ]
Bovine HapMap Consortium, 2009. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324, 528-532. [ Links ]
Brouard, J.S., Boyle, B., Ibeagha-Awemu, E.M. & Bissonnette, N., 2017. Low-depth genotyping-by-sequencing (GBS) in a bovine population: strategies to maximize the selection of high quality genotypes and the accuracy of imputation. BMC Genet. 18, 32. [ Links ]
Browning, S.R., 2008. Missing data imputation and haplotype phase inference for genome-wide association studies. Hum. Genet. 124, 439-450. [ Links ]
Browning, S.R. & Browning, B.L., 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084-1097. [ Links ]
Browning, S.R. & Browning, B.L., 2011. Haplotype phasing: Existing methods and new developments. Nat. Rev. Genet. 12, 703-714. [ Links ]
Calus, M.P.L., Bouwman, A.C., Hickey, J.M., Veerkamp, R.F. & Mulder, H.A., 2014. Evaluation of measures of correctness of genotype imputation in the context of genomic prediction: A review of livestock applications. Animal 8, 1743-1753. [ Links ]
Cleveland, M.A., Hickey, J.M. & Kinghorn, B.P., 2011. Genotype imputation for the prediction of genomic breeding values in non-genotyped and low-density genotyped individuals. BMC Proc. 5, S6. [ Links ]
Collins-Lusweti, E., 2000. Performance of Nguni, Afrikander and Bonsmara cattle under drought conditions in the North West Province of southern Africa. S. Afr. J. Anim. Sci. 30, 33-33. [ Links ]
Daetwyler, H.D., Capitan, A., Pausch, H., Stothard, P., van Binsbergen, R., Br0ndum, R.F., Liao, X., Djari, A., Rodriguez, S.C., Grohs, C., Esquerré, D., Bouchez, O., Rossignol, M-N., Klopp, C., Rocha, D., Fritz, S., Eggen, A., Bowman, P.J., Coote, D., Chamberlain, A.J., Anderson, C., VanTassell, C.P., Hulsegge, I., Goddard, M.E., Guldbrandtsen, B., Lund, M.S., Veerkamp, R.F., Boichard, D.A., Fries, R. & Hayes, B.J., 2014. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 46, 858-865. [ Links ]
Directorate: Knowledge and Information Management, 2017. Abstract of agricultural statistics. Department of Agriculture Forestry and Fisheries, Pretoria, South Africa. [ Links ]
Druet, T. & Farnir, F.P., 2011. Modeling of identity-by-descent processes along a chromosome between haplotypes and their genotyped ancestors. Genet. 188, 409-419. [ Links ]
Druet, T. & Georges, M., 2010. A hidden Markov model combining linkage and linkage disequilibrium information for haplotype reconstruction and quantitative trait locus fine mapping. Genet. 184, 789-798. [ Links ]
Druet, T., Schrooten, C. & De Roos, A.P.W., 2010. Imputation of genotypes from different single nucleotide polymorphism panels in dairy cattle. J. Dairy Sci. 93, 5443-5454. [ Links ]
Du Plessis, I., Hoffman, L.C. & Calitz, F.J., 2006. Influence of reproduction traits and pre-weaning growth rate on herd efficiency of different beef breed types in an arid sub-tropical environment. S. Afr. J. Anim. Sci. 36, 89-98. [ Links ]
Edea, Z., Dadi, H., Dessie, T., Lee, S-H. & Kim, K-S., 2015. Genome-wide linkage disequilibrium analysis of indigenous cattle breeds of Ethiopia and Korea using different SNP genotyping BeadChips. Genes Genom. 37, 759-765. [ Links ]
Ellinghaus, D., Schreiber, S., Franke, A. & Nothnagel, M., 2007. Current software for genotype imputation. Hum. Genom. 3, 371-380. [ Links ]
Elsik, C.G., Tellam, R.L. & Worley, K.C., 2009. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science 24, 522-528. [ Links ]
Falconer, D.S. & Mackay, T.F., 1996. Introduction to Quantitative Genetics. 4th edition. Longman, New York. [ Links ]
Felius, M., Beerling, M.L., Buchanan, D.S., Theunissen, B., Koolmees, P.A. & Lenstra, J.A., 2014. On the history of cattle genetic resources. Diversity 6, 705-750. [ Links ]
Ferraz, J.B.S., Wu, X., Li, H., Xu, J., Ferretti, R., Simpson, B., Walker, J., Silva, L.R., Garcia, J.F., Tait Jr., R.G. & Bauck, S., 2018. Design of a low-density SNP chip for Bos indicus: GGP indicus technical characterization and imputation accuracy to higher density SNP genotypes. In: Proceedings of the 11th World Congress of Genetics Applied to Livestock Production. Auckland, New Zealand, 6-11 February 2018. [ Links ]
Frischknecht, M., Pausch, H., Bapst, B, Signer-Hasler, H., Flury, C., Garrick, D., Stricker, C., Fries, R. & Gredler-Grandl, B., 2017. Highly accurate sequence imputation enables precise QTL mapping in Brown Swiss cattle. BMC Genomics 18, 999. [ Links ]
Fuchsberger, C., Abecasis, G.R. & Hinds, D.A., 2014. Minimac2: faster genotype imputation. Bioinf. 31, 782-784. [ Links ]
García-Ruiz, A., Ruiz-Lopez, F.J., Wiggans, G.R., Van Tassell, C.P. & Montaldo, H.H., 2015. Effect of reference population size and available ancestor genotypes on imputation of Mexican Holstein genotypes. J. Dairy Sci. 98, 3478-3484. [ Links ]
Goddard, M.E. & Hayes, B.J., 2007. Genomic selection. J. Anim. Breed. Genet. 124, 323-330. [ Links ]
Goddard, M.E., Hayes, B.J. & Meuwissen, T.H., 2010. Genomic selection in livestock populations. Genet. Res. 92, 413-421. [ Links ]
Hayes, B. & Goddard, M., 2010. Genome-wide association and genomic selection in animal breeding. Genom. 53, 876-883. [ Links ]
Hayes, B.J., Bowman, P.J., Chamberlain, A.J. & Goddard, M.E., 2009. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 92, 433-443. [ Links ]
Hayes, B.J., Bowman, P.J., Daetwyler, H.D., Kijas, J.W. & Van der Werf, J.H.J., 2012. Accuracy of genotype imputation in sheep breeds. Anim. Genet. 43, 72-80. [ Links ]
Hickey, J.M., Kinghorn, B.P., Tier, B., Wilson, J.F., Dunstan, N. & Van der Werf, J.H., 2011. A combined long-range phasing and long haplotype imputation method to impute phase for SNP genotypes. Genet. Sel. Evol. 43, 12. [ Links ]
Hickey, J.M., Crossa, J., Babu, R. & De los Campos, G., 2012. Factors affecting the accuracy of genotype imputation in populations from several maize breeding programs. Crop Sci. 52, 654-663. [ Links ]
Howie, B.N., Donnelly, P. & Marchini, J., 2009. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529. [ Links ]
Howie, B., Marchini, J. & Stephens, M., 2011. Genotype imputation with thousands of genomes. G3: Genes Genom. Genet, 1, 457-470. [ Links ]
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G.R., 2012. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955-959. [ Links ]
Hozé, C., Fouilloux, M.N., Venot, E., Guillaume, F., Dassonneville, R., Fritz, S., Ducrocq, V., Phocas, F., Boichard, D. & Croiseau, P., 2013. High-density marker imputation accuracy in sixteen French cattle breeds. Genet. Sel. Evol. 45, 33. [ Links ]
Huang, L., Wang, C. & Rosenberg, N.A., 2009. The relationship between imputation error and statistical power in genetic association studies in diverse populations. Am. J. Hum. Genet. 85, 692-698. [ Links ]
Illumina, 2006. Technical Note: 'TOP/bOt' Strand and 'A/B' Allele - A guide to Illumina's method for determining Strand and Allele for the GoldenGate® and InfiniumTM Assays. Pub. No. 370-2006-018, Available online at URL: https://www.illumina.com/documents/products/technotes/technote_topbot.pdf. [ Links ]
Judge, M.M., Kearney, J.F., McClure, M.C., Sleator, R.D. & Berry, D.P., 2016. Evaluation of developed low-density panels for imputation to higher density in independent dairy and beef cattle populations. J. Anim. Sci. 94, 949-962. [ Links ]
Khatkar, M., Nicholas, F., Collins, A., Zenger, K., Cavanagh, J., Barris, W., Schnabel, R., Taylor, J. & Raadsma, H., 2008. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genomics 9, 187. [ Links ]
Lashmar, S.F., Visser, C., Van Marle-Köster, E. & Muchadeyi, F.C., 2018. Genomic diversity and autozygosity within the SA Drakensberger beef cattle breed. Livest. Sci. 212, 111-119. [ Links ]
Li, Y., Willer, C., Sanna, S. & Abecasis, G., 2009. Genotype imputation. Ann. Rev. Genom. Hum. Genet. 10, 387-406. [ Links ]
Li, Y., Willer, C.J., Ding, J., Scheet, P. & Abecasis, G.R., 2010. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 34, 816-834. [ Links ]
Lin, P., Hartz, S.M., Zhang, Z.H., Saconne, S.F., Wang, J., Tischfield, J.A., Edenberg, H.J., Kramer, J.R., Goate, A.M., Bierut, L.J. & Rice, J.P., 2010. A new statistic to evaluare imputation reliability. PloS one 5, e9697. [ Links ]
Ma, P., Br0ndum, R.F., Zhang, Q., Lund, M.S. & Su, G., 2012. Comparison of different methods for imputing genome-wide marker genotypes in Swedish and Finnish red cattle. J. Dairy Sci. 96, 4666-4677. [ Links ]
Makina, S.O., Muchadeyi, F.C., Van Marle-Köster, E., MacNeil, M.D. & Maiwashe, A., 2014. Genetic diversity and population structure among six cattle breeds in South Africa using a whole genome SNP panel. Front. Genet. 5, 1-7. [ Links ]
Makina, S.O., Taylor, J.F., Van Marle-Köster, E., Muchadeyi, F.C., Makgahlela, M.L., MacNeil, M.D. & Maiwashe, A., 2015a. Extent of linkage disequilibrium and effective population size in four South African Sanga cattle breeds. Front. Genet. 6, 1-12. [ Links ]
Makina, S.O., Muchadeyi, F.C., Van Marle-Köster, E., Taylor, J.F., Makgahlela, M.L. & Maiwashe, A., 2015b. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 47, 92. [ Links ]
Makina, S.O., Whitacre, L.K., Decker, J.E., Taylor, J.F., MacNeil, M.D., Scholtz, M.M., Van Marle-Köster, E., Muchadeyi, F.C., Makgahlela, M.L. & Maiwashe, A., 2016. Insight into the genetic composition of South African Sanga cattle using SNP data from cattle breeds worldwide. Genet. Sel. Evol. 48, 88. [ Links ]
Mapholi, N.O., Maiwashe, A., Matika, O., Riggio, V., Bishop, S.C., MacNeil, M.D., Banga, C., Taylor, J.F. & Dzama, K., 2016. Genome-wide association study of tick resistance in South African Nguni cattle. Ticks Tick-Borne Dis. 7, 487-497. [ Links ]
Marchini, J. & Howie, B., 2010. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499-511. [ Links ]
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P., 2007. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39, 906-913. [ Links ]
Matukumalli, L.K., Lawley, C.T., Schnabel, R.D., Taylor, J.F., Allan, M.F., Heaton, M.P., O'Connell, J., Moore, S.S., Smith, T.P., Sonstegard, T.S. & Van Tassell, C.P., 2009. Development and characterization of a high density SNP genotyping assay for cattle. PLoS one 4, e5350-5063. [ Links ]
Maule, J.P., 1973. The role of the indigenous breeds for beef production in southern Africa. S. Afr. J. Anim. Sci. 3, 111-132. [ Links ]
Meissner, H.H., Scholtz, M.M. & Palmer, A.R., 2013. Sustainability of the South African livestock sector towards 2050 Part 1: Worth and impact of the sector. S. Afr. J. Anim. Sci. 43, 282-297. [ Links ]
Meuwissen, T.H.E., Hayes, B.J. & Goddard, M.E., 2001. Prediction of total genetic value using genome-wide dense marker maps. Genet. 157, 1819-1829. [ Links ]
Milanesi, M., Vicario, D., Stella, A., Valentini, A., Ajmone - Marsan, P.A.O.L.O., Biffani, S., Biscarini, F., Jansen, G. & Nicolazzi, E.L., 2015. Imputation accuracy is robust to cattle reference genome updates. Anim. Genet. 46, 69-72. [ Links ]
Minozzi, G., Williams, J.L., Stella, A., Strozzi, F., Luini, M., Settles, M.L., Taylor, J.F., Whitlock, R.H., Zanella, R. & Neibergs, H.L., 2012. Meta-analysis of two genome-wide association studies of bovine paratuberculosis. PLoS one 7, e32578. [ Links ]
Mokry, F.B., Buzanskas, M.E., de Alvarenga Mudadu, M., do Amaral Grossi, D., Higa, R.H., Ventura, R.V., de Lima, A.O., Sargolzaei, M., Meirelles, S.L.C., Schenkel, F.S., da Silva, M.V.G.B., Niciura, S.C.M., de Alencar, M.M., Munari, D.P. & de Almeida Regitano, L.C., 2014. Linkage disequilibrium and haplotype block structure in a composite beef cattle breed. BMC Genomics 15, S6. [ Links ]
Mulder, H.A., Calus, M.P.L., Druet, T. & Schrooten, C., 2012. Imputation of genotypes with low-density chips and its effect on reliability of direct genomic values in Dutch Holstein cattle. J. Dairy Sci. 95, 876-889. [ Links ]
Nicolazzi, E.L., Biffani, S. & Jansen, G., 2013. Imputing genotypes using PedImpute fast algorithm combining pedigree and population information. J. Dairy Sci. 96, 2649-2653. [ Links ]
Nicolazzi, E.L., Biffani, S., Biscarini, F., Orozco ter Wengel, P., Caprera, A., Nazzicari, N. & Stella, A., 2015. Software solutions for the livestock genomics SNP array revolution. Anim. Genet. 46, 343-353. [ Links ]
O'Connell, J., Gurdasani, D., Delaneau, O., Pirastu, N., Ulivi, S., Cocca, M., Traglia, M., Huang, J., Huffman, J.E., Rudan, I. & McQuillan, R., 2014. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10, e1004234. [ Links ]
Ogawa, S., Matsuda, H., Taniguchi, Y., Watanabe, T., Takasuga, A., Sugimoto, Y. & Iwaisaki, H., 2016. Accuracy of imputation of single nucleotide polymorphism marker genotypes from low- density panels in Japanese Black cattle. Anim. Sci. J. 87, 3-12. [ Links ]
Pausch, H., Aigner, B., Emmerling, R., Edel, C., Götz, K.U. & Fries, R., 2013. Imputation of high-density genotypes in the Fleckvieh cattle population. Genet. Sel. Evol. 45, 3. [ Links ]
Pausch, H., MacLeod, I.M., Fries, R., Emmerling, R., Bowman, P.J., Daetwyler, H.D. & Goddard, M.E., 2017. Evaluation of the accuracy of imputed sequence variant genotypes and their utility for causal variant detection in cattle. Genet. Sel. Evol. 49, 24. [ Links ]
Payne, W.J.A. & Hodges, J., 1997. Tropical cattle: origins, breeds and breeding policies. Blackwell Science Ltd., Oxford, UK. [ Links ]
Pei, Y.F., Li, J., Zhang, L., Papasian, C.J. & Deng, H.W., 2008. Analyses and comparison of accuracy of different genotype imputation methods. PloS one 3, e3551. [ Links ]
Phuong, T.M., Lin, Z. & Altman, R.B., 2006. Choosing SNPs using feature selection. J. Bioinform. Comput. Biol. 4, 241-257. [ Links ]
Piccoli, M.L., Braccini, J., Cardoso, F.F., Sargolzaei, M., Larmer, S.G. & Schenkel, F.S., 2014. Accuracy of genome-wide imputation in Braford and Hereford beef cattle. BMC Genet. 15, 157. [ Links ]
Pienaar, L., Grobler, J.P., Neser, F.W.C., Scholtz, M.M., Swart, H., Ehlers, K. & Marx, M., 2014. Genetic diversity in selected stud and commercial herds of the Afrikaner cattle breed. S. Afr. J. Anim. Sci. 44, 80-84. [ Links ]
Porter, V., 1991. Cattle. A Handbook to the Breeds of the World. Christopher Helm, London. pp. 145-146. [ Links ]
Pritchard, J.K. & Cox, N.J., 2002. The allelic architecture of human disease genes: common disease-common variant...or not?. Hum. Mol. Genet. 11, 2417-2423. [ Links ]
Purcell, S., Neale, B. & Todd-Brown, K., 2007. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559-575. [ Links ]
Purfield, D.C., McClure, M. & Berry, D.P., 2016. Justification for setting the individual animal genotype call rate threshold at eighty-five percent. J. Anim. Sci. 94, 4558-4569. [ Links ]
Qanbari, S., Pimentel, E.C., Tetens, J., Thaller, G., Lichtner, P., Sharifi, A.R. & Simianer, H., 2010. The pattern of linkage disequilibrium in German Holstein cattle. Anim. Genet. 41, 346-356. [ Links ]
Qwabe, S.O., Van Marle-Köster, E., Maiwashe, A. & Muchadeyi, F.C., 2013. Evaluation of the BovineSNP50 genotyping array in four South African cattle populations. S. Afr. J. Anim. Sci. 43, 64-67. [ Links ]
Rabiner, L., 1989. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE 77, 257-286. [ Links ]
Ramnarine, S., Zhang, J., Chen, L.S., Culverhouse, R., Duan, W., Hancock, D.B., Hartz, S.M., Johnson, E.O., Olfson, E., Schwantes-An, T.H. & Saccone, N.L., 2015. When does choice of accuracy measure alter imputation accuracy assessments? PloS one 10, e0137601. [ Links ]
Rechav, Y. & Kostrzewski, M.W., 1991. The relative resistance of six cattle breeds to the tick Boophilus decoloratus in South Africa. Onderstepoort J. Vet. Res. 58, 181-186. [ Links ]
Rosen, B.D., Bickhart, D.M., Schnabel, R.D., Koren, S., Elsik, C.G., Zimin, A., Dreischer, C., Schultheiss, S., Hall, R., Schroeder, S.G., Van Tassell, C.P., Smith, T.P.L. & Medrano, J.F., 2018. Modernizing the bovine reference genome assembly. In: Proceedings of the 11th World Congress of Genetics Applied to Livestock Production. Auckland, New Zealand, 6-11 February 2018. [ Links ]
Roshyara, N.R., Kirsten, H., Horn, K., Ahnert, P. & Scholz, M., 2014. Impact of pre-imputation SNP-filtering on genotype imputation results. BMC Genet. 15, 88. [ Links ]
Rutkoski, J.E., Poland, J., Jannink, J.-L. & Sorrells, M.E., 2013. Imputation of unordered markers and the impact on genomic selection accuracy. G3 Genes Genomes Genet. 3, 427-439. [ Links ]
SA Stud Book, 2016. SA Stud Book annual report. Available online at: http://www.sastudbook.co.za/images/photos/Annual_Report_2016_a.pdf. [ Links ]
SA Stud Book, 2017. Media Release: Genomic Breeding Values for Bonsmara. Available online at: http://www.sastudbook.co.za/n16/general-news/media-release:-genomic-breeding-values-for-bonsmara.html. [ Links ]
Sargolzaei, M., Chesnais, J.P. & Schenkel, F.S., 2014. A new approach for efficient genotype imputation using information from relatives. BMC Genomics 15, 478. [ Links ]
Scheet, P. & Stephens, M., 2006. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78, 629-644. [ Links ]
Scholtz, M.M., 1988. Selection possibilities of hardy beef breeds in Africa: The Nguni example. In: Proceedings of the 3rd World Congress on Sheep and Beef Cattle Breeds. Paris, France, 19-23 June 1988. pp. 303-319. [ Links ]
Scholtz, M.M., 2010. Beef breeding in South Africa. 2nd edition. Pretoria, South Africa. [ Links ]
Scholtz, M.M., McManus, G., Leeuw, K-C., Louvandini, H., Seixas, L., Demelo, C.B., Theunissen, A. & Neser, F.W.C., 2013. The effect of global warming on beef production in developing countries of the southern hemisphere. Nat. Sci. 5, 106-119. [ Links ]
Scholtz, M.M., Maiwashe, A., Neser, F.W.C., Theunissen, A., Olivier, W.J., Mokolobate, M.C. & Hendriks, J., 2014. Livestock breeding for sustainability to mitigate global warming, with the emphasis on developing countries. S. Afr. J. Anim. Sci. 43, 269-281. [ Links ]
Schoeman, S.J., 1989. Recent research into the production potential of indigenous cattle with special reference to the Sanga. S. Afr. J. Anim. Sci. 19, 55-61. [ Links ]
Schrooten, C., Dassonneville, R., Ducrocq, V., Br0ndum, R.F., Lund, M.S., Chen, J., Liu, Z., González-Recio, O., Pena, J. & Druet, T., 2014. Error rate for imputation from the Illumina BovineSNP50 chip to the Illumina BovineHD chip. Genet. Sel. Evol. 46, 10. [ Links ]
South African Weather Service, 2016. South African Weather Service Annual Report 2016/2017. Available online at: https://nationalgovernment.co.za/entity_annual/1364/2017-south-african-weather-service-annual-report.pdf. [ Links ]
Spencer, C.C., Su, Z., Donnelly, P. & Marchini, J., 2009. Designing genome-wide association studies: Sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 5, e1000477. [ Links ]
Statistics South Africa, 2018. Statistical release P0302: Mid-year population estimates 2018. Available online at: https://www.statssa.gov.za/publications/P0302/P03022018.pdf. [ Links ]
Strydom, P.E., 2008. Do indigenous southern African cattle breeds have the right genetics for commercial production of quality meat? Meat Sci. 80, 86-93. [ Links ]
Tsai, H.Y., Matika, O., Edwards, S.M., Antolín-Sánchez, R., Hamilton, A., Guy, D.R., Tinch, A.E., Gharbi, K., Stear, M.J., Taggart, J.B. & Bron, J.E., 2017. Genotype imputation to improve the cost-efficiency of genomic selection in farmed Atlantic salmon. G3: Genes Genom. Genet. 7, 1377-1383. [ Links ]
United Nations, 2017. World population prospects: The 2017 revision. Volume I: Comprehensive tables (ST/ESA/SER.A/399). Department of Economic and Social Affairs, Population Division. Available online at: https://population.un.org/wpp/Publications/Files/WPP2017_Volume-I_Comprehensive-Tables.pdf. [ Links ]
Van Binsbergen, R., Bink, M.C., Calus, M.P., Van Eeuwijk, F.A., Hayes, B.J., Hulsegge, I. & Veerkamp, R.F., 2014. Accuracy of imputation to whole-genome sequence data in Holstein Friesian cattle. Genet. Sel. Evol. 46, 41. [ Links ]
Van der Westhuizen, R.R., Van Marle-Köster, E., Theron, H.E. & Van der Westhuizen, J., 2014. Reference population for South African Bonsmara cattle. In: Proceedings of the 10th World Congress of Genetics Applied to Livestock Production. Vancouver, Canada, 17-22 August 2014. Poster presentation 498. [ Links ]
Van Marle, J., 1974. The breeding of beef cattle in South Africa: Past, present and future. S. Afr. J. Anim. Sci. 4, 297-304. [ Links ]
Van Marle-Köster, E. & Visser, C., 2018. Genetic improvement in South African livestock: Can genomics bridge the gap between the developed and developing sectors? Front. Genet. 9, 1-12. [ Links ]
Van Marle-Köster, E., Visser, C. & Berry, D.P., 2013. A review of genomic selection - Implications for the South African beef and dairy cattle industries. S. Afr. J. Anim. Sci. 43, 1-17. [ Links ]
Van Raden, P.M. & Sun, C., 2014. Fast imputation using medium- or low-coverage sequence data. In: Proceedings of the 10th World Congress of Genetics Applied to Livestock Production. Vancouver, Canada, 17-22 August 2014. Comm. 179. [ Links ]
Ventura, R.V., Lu, D., Schenkel, F.S., Wang, Z., Li, C. & Miller, S.P., 2014. Impact of reference population on accuracy of imputation from 6K to 50K single nucleotide polymorphism chips in purebred and crossbreed beef cattle. J. Anim. Sci. 92, 1433-1444. [ Links ]
Ventura, R.V., Miller, S.P., Dodds, K.G., Auvray, B., Lee, M., Bixley, M., Shannon, M.C. & McEwan, J.C., 2016. Assessing accuracy of imputation using different SNP panel densities in a multi-breed sheep population. Genet. Sel. Evol. 48, 71. [ Links ]
Verma, S.S., De Andrade, M., Tromp, G., Kuivaniemi, H., Pugh, E., Namjou-Khales, B., Mukherjee, S., Jarvik, G.P., Kottyan, L.C., Burt, A. & Bradford, Y., 2014. Imputation and quality control steps for combining multiple genome-wide datasets. Front. Genet. 5, 370. [ Links ]
Wang, M.D., Dzama, K., Hefer, C.A. & Muchadeyi, F.C., 2015. Genomic population structure and prevalence of copy number variations in South African Nguni cattle. BMC Genomics 16, 894. [ Links ]
Wang, Y., Lin, G., Li, C. & Stothard, P., 2016. Genotype imputation methods and their effects on genomic predictions in cattle. Springer Sci. Rev. 4, 79-98. [ Links ]
Weigel, K.A., Van Tassell, C.P., O'Connell, J.R., VanRaden, P.M. & Wiggans, G.R., 2010. Prediction of unobserved single nucleotide polymorphism genotypes of Jersey cattle using reference panels and population-based imputation algorithms. J. Dairy Sci. 93, 2229-2238. [ Links ]
Wellmann, R., Preuß, S., Tholen, E., Heinkel, J., Wimmers, K. & Bennewitz, J., 2013. Genomic selection using low density marker panels with application to a sire line in pigs. Genet. Sel. Evol. 45, 28. [ Links ]
Wiggans, G.R., Cooper, T.A., Van Tassell, C.P., Sonstegard, T.S. & Simpson, E.B., 2013. Technical note: Characteristics and use of the Illumina BovineLD and GeneSeek Genomic Profiler low-density bead chips for genomic evaluation. J. Dairy Sci. 96, 1258-1263. [ Links ]
Wu, X.L., Xu, J., Feng, G., Wiggans, G.R., Taylor, J.F. & Bauck, S., 2016. Optimal design of low-density SNP arrays for genomic prediction: Algorithm and applications. PloS one 11, e0161719. [ Links ]
Yuan, X., Zhang, J. & Wang, Y., 2011. Simulating linkage disequilibrium structures in a human population for SNP association studies. Biochem. Genet. 49, 395-409. [ Links ]
Zhang, Z. & Druet, T., 2010. Marker imputation with low-density marker panels in Dutch Holstein cattle. J. Dairy Sci. 93, 5487-5494. [ Links ]
Zimin, A.V., Delcher, A.L., Florea, L., Kelley, D.R., Schatz, M.C., Puiu, D., Hanrahan, F., Pertea, G., Van Tassell, C.P., Sonstegard, T.S., Marçais, G., Roberts, M., Subramanian, P., Yorke, J.A. & Salzberg, S.L., 2009. A whole-genome assembly of the domestic cow, Bos Taurus. Genome Biol. 10, R42. [ Links ]
Zwane, A.A., 2017. Genome-wide marker discovery in three South African indigenous cattle breeds (Afrikaner, Drakensberger and Nguni) using whole genome sequencing. PhD thesis, University of Pretoria, November 2017. [ Links ]
Zwane, A.A., Maiwashe, A., Makgahlela, M.L., Choudhury, A., Taylor, J.F. & Van Marle-Köster, E., 2016. Genome-wide identification of breed-informative single-nucleotide polymorphisms in three South African indigenous cattle breeds. S. Afr. J. Anim. Sci. 46, 302-312. [ Links ]
Received 21 May 2018
Accepted 17 February 2019
First published online 11 April 2019
# Corresponding author: s.lashmar@gmail.com


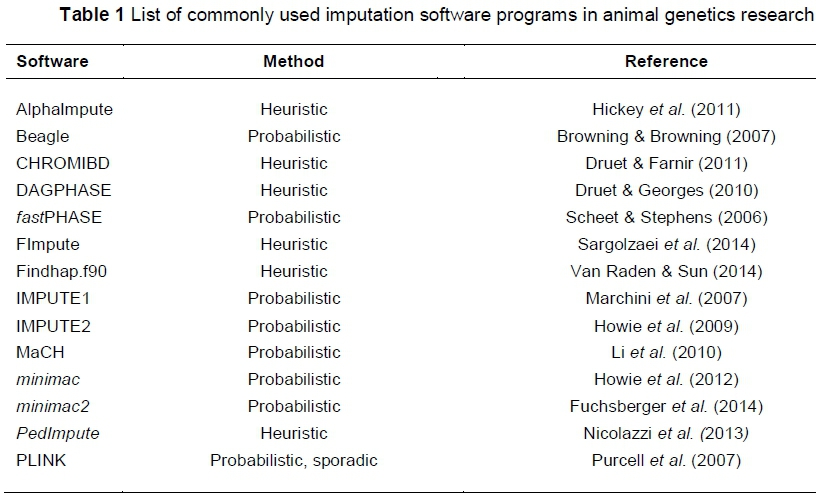{kind=link}
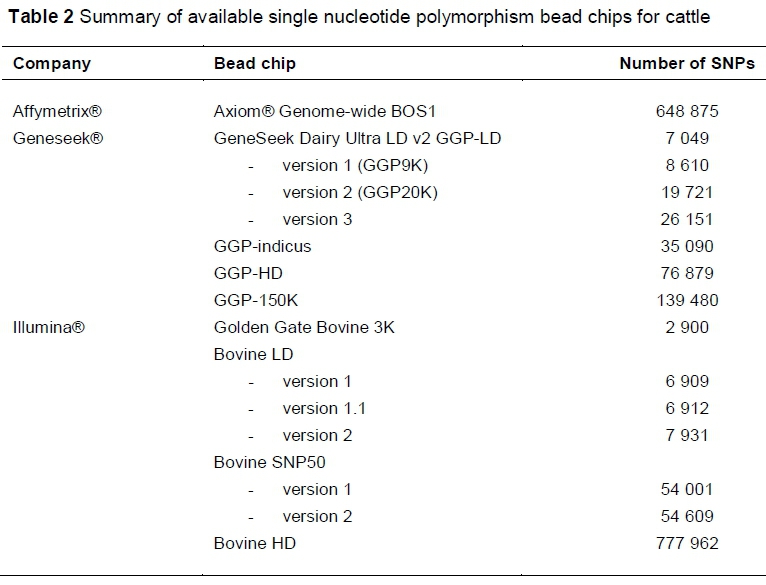{kind=link}