










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Animal Science
On-line version ISSN 2221-4062
Print version ISSN 0375-1589
S. Afr. j. anim. sci. vol.49 n.1 Pretoria 2019
http://dx.doi.org/10.4314/sajas.v49i1.11
ARTICLES
Investigation of single nucleotide polymorphisms in porcine candidate genes for blood component traits in pigs
B. Lim; S. Kim; K. Do#
Department of Animal Biotechnology, Faculty of Biotechnology, Jeju National University, Jeju 63243, Republic of Korea
ABSTRACT
This study used 209 public single nucleotide polymorphism (SNP) arrays for 151 candidate genes of pigs to analyse their association with nine blood component traits (insulin-like growth factor-I, insulin, immuno globulin, lymphocyte, monocyte, eosinophil, basophil, neutrophil and atypical lymph) in 209 Korean native pigs and Yorkshire F2 hybrids. Of these, 52 SNPs in 49 candidate genes showed significant association with one or more blood component traits. Nineteen of these SNPs were found to be present in blood component QTL regions. The 49 candidate genes corresponding to 52 SNPs with significant effects were detected and used for gene ontology analysis to understand the function of the candidate genes at molecular level. Based on functional classification (biological process, cellular components, and molecular function) of annotated candidates, 34 candidate genes (11 genes of IGF-1, 9 of IS, 9 of IG, 6 of NP, and 3 of EP) were detected. Additionally, eight genes (PSMB4, PSME3, MAPKAPK3, CTLA4, CUL7, GGT1, IDH3B, and RXRB) interacting with four immune pathways (immune system, adaptive immune system, Class I MHC-mediated antigen processing and presentation, and antigen processing: ubiquitination and proteasome degradation) were found through pathway and network analyses. The eight candidate genes identified in this study are included in class I MHC-mediated antigen pathway, which is an important factor that determines the success of organ transplantation in addition to the improvement of diseases and immunity of pigs. Therefore, these genes can potentially be used in heterogeneous organ research in future research.
Keywords: Association analysis, gene network, pathway, SNP, swine
Introduction
For the success of the swine industry, it is important to produce safe high-quality pigs and prevent or reduce the incidence of infectious diseases in pigs. Although the development of livestock breeding has been greatly improved through many studies on growth and meat quality domestically and abroad, there have been insufficient numbers of recent studies on the improvement of traits related to diseases and immunity worldwide. In particular, modern pig farming is a large and intensive production system, and the pigs are easily exposed to pathogens that cause infectious diseases and pathologies. As the incidence of diseases worldwide increases, studies are being conducted to improve the capacity of the pig immune system (Schroyen & Tuggle, 2015). In addition, pigs are useful as human biomedical models and in genetic studies that involve the immune system, because they are highly comparable with humans in terms of their anatomy, physiology, genetics and genomics (Lunney, 2007).
Single nucleotide polymorphisms (SNPs) are simple genetic characters that are widely distributed and are the most abundant type of variation in the whole genome. SNPs have the advantages of relatively high availability and stability compared with microsatellites and other DNA polymorphism markers. Therefore, SNPs are important markers that can be applied in various fields (e.g. evolution and disease) of the study of livestock (Kijas et al., 2009).
Previous studies related to pig disease that used SNP markers have shown that the WUR10000125 single-base mutation in the GBP1 (guanylate binding protein 1) gene located on pig chromosome 4, and c.2509 G>C, c.2638 G>A, and c.3534 C>T mutations in the CD163 (cluster of differentiation 163) gene on chromosome 5 significantly affect viremia and weight gain in serum when pigs are infected with porcine reproductive and respiratory syndrome virus (PRRSV) (Boddicker et al., 2012; Lim et al., 2018). In addition, the ALGA1150315 single-base mutation located on pig chromosome 9 and the ALGA0122080 and MARC0001766 mutations located on chromosome 12 have been reported to have significant effects on the same traits in pigs that are infected with porcine circovirus (PCV) (McKnite et al., 2014).
Recent research using these methods has identified critical candidate genes through gene network and pathway analysis in immune response experiments related to PRRSV resistance and gram-negative Bacillus resistance in pigs (Wysocki et al., 2012; Zhao et al., 2012).
This study was implemented to carry out an association analysis of blood component traits to 209 public SNP arrays of 151 candidate genes in Korean native pigs (KNP) and Yorkshire F2 hybrid populations. The authors also carried out a gene ontology (GO) analysis to identify the potential role of these candidate genes in metabolism and network and pathway analyses of gene interactions to detect candidate genes that are related to the immune function in pigs.
Materials and Methods
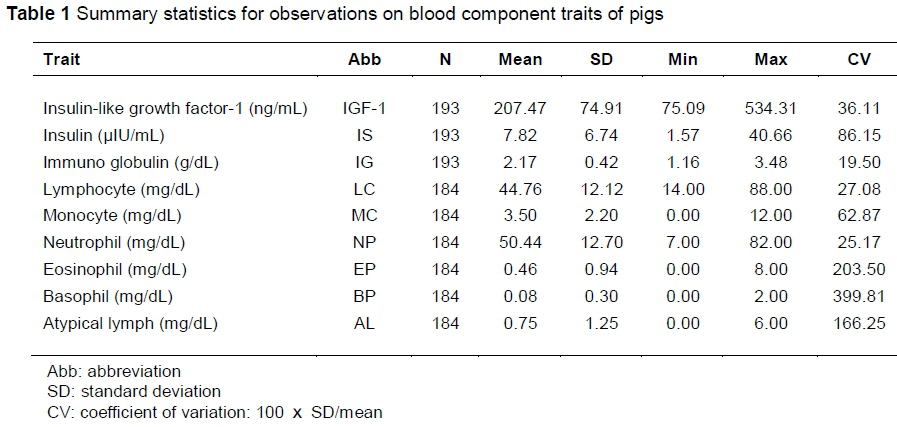
The pig population used in this study was a sample of 209 KNP and Yorkshire F2 hybrid groups, which had been used for predecessor QTL discovery, SNP association, and candidate gene identification studies (Kim et al., 2011; Lee et al., 2012). In the F2 group, levels of nine blood components that were related to immunity were measured in serum: insulin-like growth factor-1 (IGF-1), insulin (IS), immuno globulin (IG), lymphocyte (LC), monocyte (MC), eosinophil (EP), basophil (BP), neutrophil (NP) and atypical lymph (AL). Table 1 shows the mean values and their standard deviation for each of these blood component traits.
The SNPs used in this experiment were 209 confirmed SNPs, from which 22 breed-specific SNPs were removed. Thirty SNPs among the 261 SNPs selected by Li et al. (2011) were also excluded, which were not mapped to pig genome build 11 using the NCBI blast tool. The genotypes of the confirmed SNPs were analysed by a MassARRAY method using the MassARRAY Design Software (Sequenom Inc, San Diego, USA) (Li et al., 2011). Amplification primers were designed using Spectro DESIGNER 1.3.4 (Sequenom Inc, San Diego, USA) and SNPs were multiplexed in 14 assays using the manufacturer's instructions for the iPLEX system (Sequenom Inc, San Diego, USA).
Quality control (QC) of the 209 SNP markers that were analysed by genotyping was carried out using the PLINK 1.07 programme. An SNP was removed based on two criteria, minor allele frequency (MAF) <0.05 and call rate <50% (Purcell et al., 2007). To estimate the linkage disequilibrium (LD) between mutations in genes on the same chromosome, analysis was performed using the Haploview software package (Barrett et al., 2004) and the values of D' and r2 were estimated using the methods of Stephens et al. (2001). Mutations with r2 > 0.8 and LD values within the range of those of chromosomal single-base mutations were selected, and then used in the association analysis (Barrett et al., 2004).
To evaluate the genotype effects of the selected SNP markers by QC and LD analyses, association analysis was performed using the generalized linear model (GLM) function of SAS 9.4 software package. The model used for the analysis was as follows:
Yijklm = μ + Si + Gj+ b1Ak+ eiJkl
where: Yijklmis the observed value of blood component traits μ is the mean of the samples Siis the effect of sex Gjis the effect of genotype Al is the covariate of slaughter age (days) b1is the regression coefficient of slaughter age (days eijklis the random error
The candidate genes containing SNPs that have significant effects on blood component traits were selected through association and GO analyses using the functional annotation tool provided by the database for annotation, visualization, and integrated discovery (DAVID). In the GO analysis, the relevance of candidate genes was determined by classifying them in three functional groups (biological process, cellular components and molecular function), based on their associations.
Gene pathway and network analyses provide crucial insight into the genetic structure behind complex polygenic traits (Kadarmideen et al., 2006). Therefore, gene pathway and network analyses were performed in the GeneMANIA and KEGGParser tool plugins, respectively, supported by Cytoscape software 3.6.1 for candidate genes based on blood component traits-related GO term results (Shannon et al., 2003).
Results
Sixty-five SNPs of the 209 public SNPs examined were filtered out by QC and LD, leaving 144 SNPs for further analyses. Forty-one of the 65 filtered SNPs were removed through further QC. Twenty-four SNPs had a MAF lower than 0.05 and 18 had a call rate lower than 50%. The remaining 24 SNPs of these 65 were eliminated during LD analysis because their r2 values were not higher than 0.8. Finally, the association of the 144 SNPs filtered through QC and LD with blood component traits was analysed.
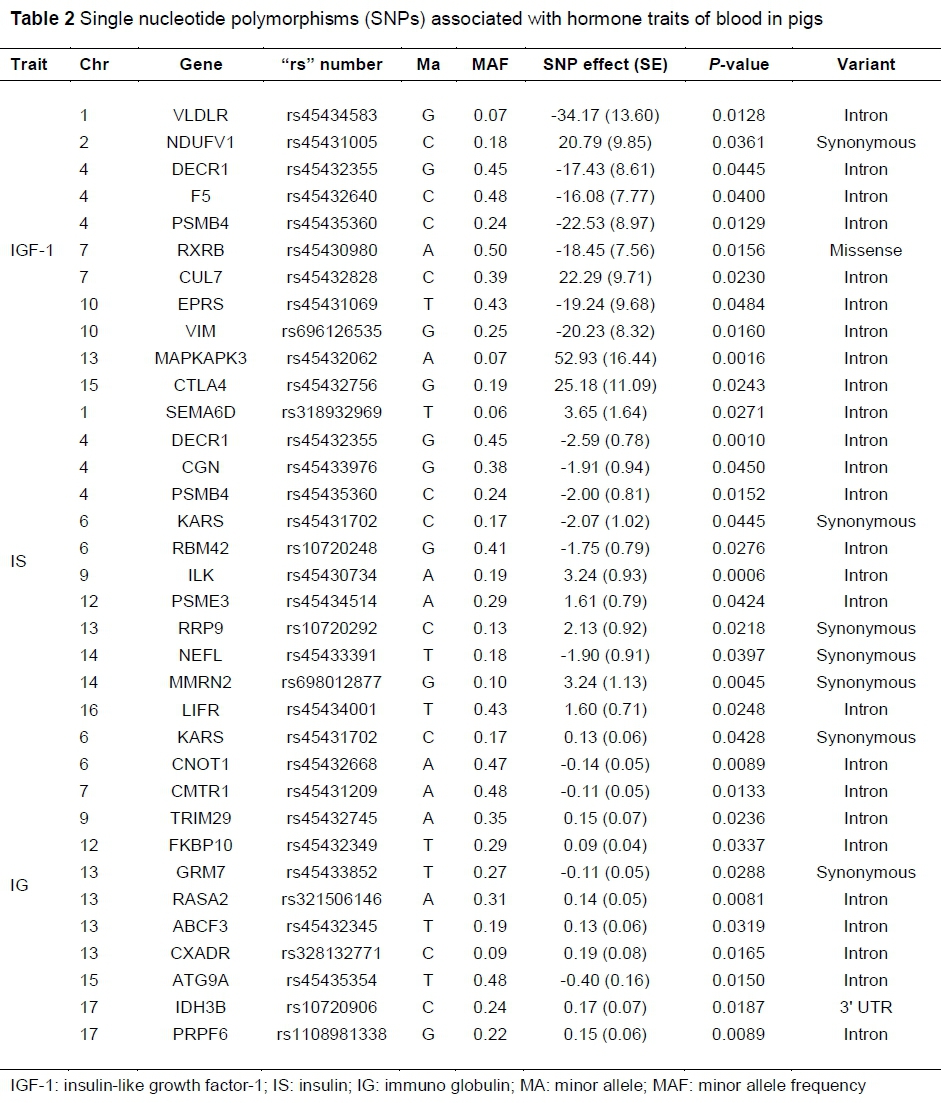
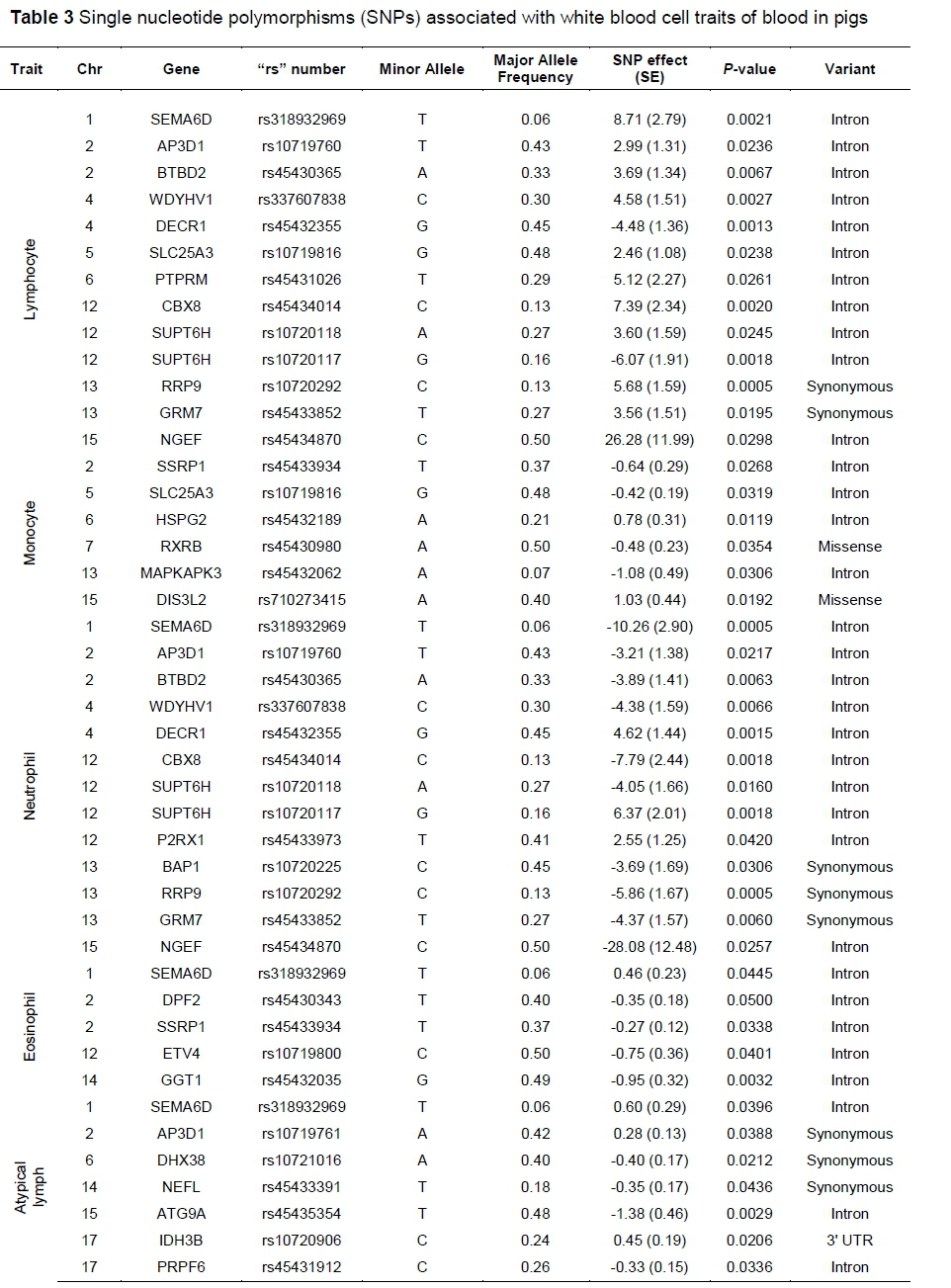
An association analysis was performed between the 144 filtered SNPs and nine blood component trait (IGF-1, IS, IG, LC, MC, NP, EP, BP, and AL), and significantly associated SNPs were detected (Table 2; Table 3). A total of 52 SNPs showed significant associations with one or more of the blood component traits. The SNPs identified in the association analysis were classified as 2 missense variants, 9 synonymous variants, 1 UTR variant and 40 intron variants. Among these, 4 SNPs - including the SNP rs318932969 (associated with IS, LC, NP, EP, and AL) located in the SEMA6D gene, rs45432355 (associated with IGF-1, IS, LC, and NP) in the DECR1 gene, rs45433852 (associated with IG, LC, and NP) in the GRM7 gene, and rs10720292 (associated with IS, LC, and NP) in the RRP9 gene - were found to be significantly associated with three or more blood component traits. Based on the blood component traits, 11 SNPs were significantly associated with IGF-1, 12 with IS, 12 with IG, 13 with LC, 6 with MC, 13 with NP, 5 with EP, and 7 with AL trait (all P <0.05).
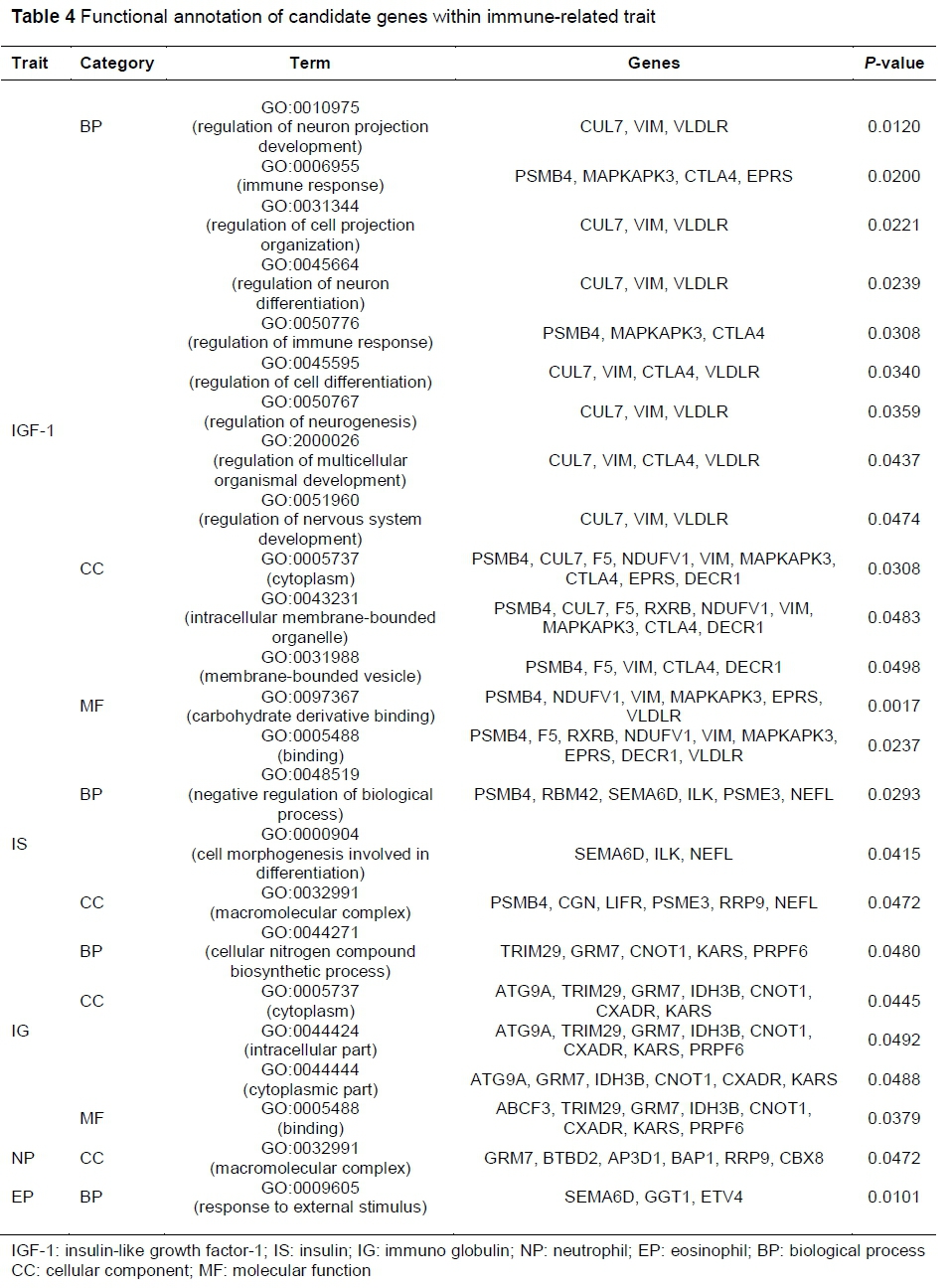
Genes containing those SNPs that showed significant associations with blood component traits were selected as candidate genes and used for GO analysis. A total of 38 candidate genes were detected, 11 (CUL7, VIM, VLDLR, PSMB4, MAPKAPK3, CTLA4, EPRS, F5, NDUFV1, DECR1 and RXRB) of which were related with IGF-1 ; 9 (PSMB4, RBM42, SEMA6D, ILK, PSME3, NEFL, CGN, LIFR and RRP9) with IS; 9 (TRIM29, GRM7, CNOT1, KARS, PRPF6, ATG9A, IDH3B, CXADR and ABCF3) with IG; 6 (GRM7, BTBD2, AP3D1, BAP1, RRP9 and CBX8) with NP; and 3 (SEMA6D, GGT1 and ETV4) were related with EP. Four (PSMB4, SEMA6D, GRM7 and RRP9) of these genes overlapped between two traits. The 11 candidate genes related with IGF-1 trait were detected in 14 GO terms (9 biological processes, 3 cellular components and 2 molecular functions), 9 genes of IS were detected in 3 GO terms (2 biological processes and 1 cellular component), 9 genes of IG were detected in 5 GO terms (1 biological process, 3 cellular components and 1 molecular function), 6 genes of NP were detected in 1 GO term (a cellular component), and 3 genes of EP were detected in 1 GO term (a biological process) (Table 4).
Pathway searches were performed on selected candidate genes using the GeneMANIA and KEGGParser analysis tools, and the direct interactions between candidate genes were also investigated.
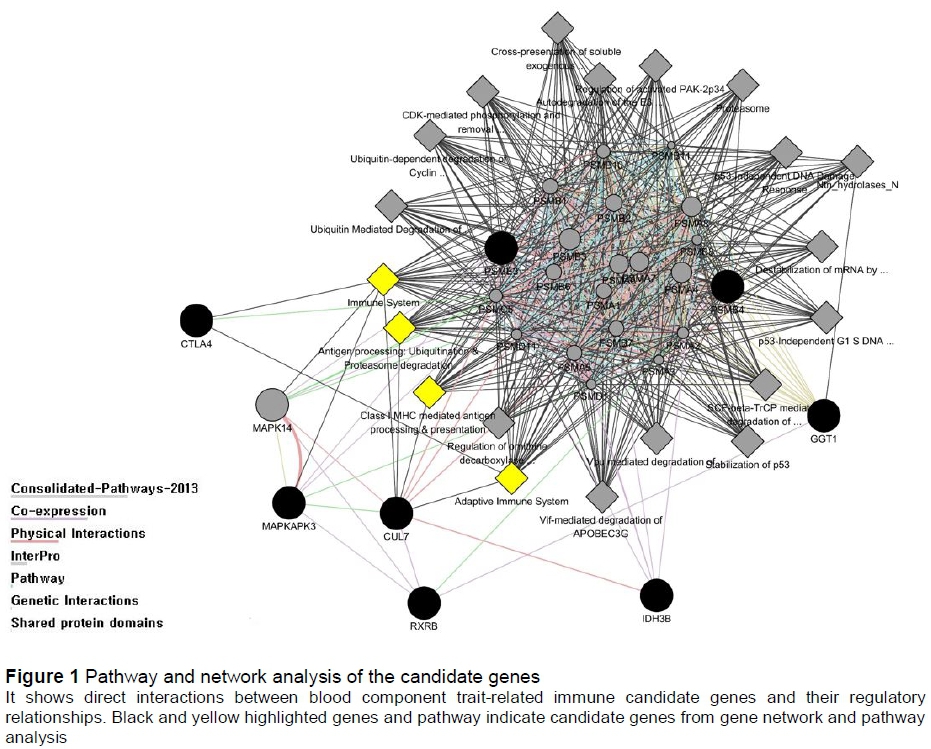
Eight genes (PSMB4, PSME3, MAPKAPK3, CTLA4, CUL7, GGT1, IDH3B, and RXRB) had integrated interactions in four blood component trait-related immune pathways (immune system, adaptive immune system, class I MHC-mediated antigen processing and presentation, and antigen processing: ubiquitination and proteasome degradation), and genetic interactions among candidate genes were also observed (Figure 1).
Discussion
This study was conducted to investigate blood component traits-related immune candidate genes using 209 publicly available SNPs of 151 genes in KNP and Yorkshire F2 hybrid pig populations.
The genotypes of 209 SNPs were analysed using the Sequenom MassARRAY system method, which is the method that the authors used in a previous study (Li et al., 2011). Using 144 SNPs filtered through QC and LD analyses, they performed association analysis on nine blood component traits (IGF-1, IS, IG, LC, MC, EP, BP, NP and AL) and hormones.
Table 2 and Table 3 show that 52 SNPs among the 144 SNPs were effectively associated with 9 blood component traits, and 19 SNPs were included in blood component traits-related immune QTL regions. In previous reports, the SNPs in the QTL region of genes related to PRRSV susceptibility (123,334-124,643 kb) and PRRS viral load (32,901-33,123 kb) were SEMA6D (rs318932969), IDH3B (rs10720906), and HSPG2 (rs45432189) (Boddicker et al., 2012; Waide et al., 2017). In the QTL regions (609-21,136 kb) of the interleukin 10 level and Toll-like receptor 9 level genes that affect the resistance to viral disease (Uddin et al., 2011), the SNPs NDUFV1, DPF2, and SSRP1, were found on chromosome 2. In addition, the CSFV antibody level and CD4-positive/CD8-positive leukocyte ratio QTL regions (80,511-106,510 kb) related to classical swine fever (CSF) virus infection were found to contain the CGN and PSMB4 SNPs located on chromosome 4. Further, the CD4-positive/CD8-positive leukocyte ratio QTL region (57,165-127,880 kb) was also reported to be present on chromosome 15, and SNPs were observed as CTLA4 (rs45432756) and ATG9A (rs45436465) in the same region (Lu et al., 2011; Lu et al., 2014). Moreover, the CD4-negative and CD8-positive leukocyte percentage QTLs, which are related to the disease resistance of pigs (Lu et al., 2011), were found on chromosome 13 (27,210-58,927 kb), and MAPKAPK3 (rs45432062), RRP9 (rs10720292), and BAP1 (rs10720225) SNPs were present in the same QTL region. The QTL region of genes that are associated with traits for Salmonella count in the liver, and in the liver and spleen, which were reported to be associated with porcine salmonellosis susceptibility, showed that AP3D1 (rs10719761) and BTBD2 (rs45430365) SNPs on chromosome 2 (75,040-121,864 kb) were located within the QTL regions (Galina-Pantoja et al., 2009). In addition, the SNPs PTPRM (rs45431026) located on chromosome 6, SUPT6H (rs10720118) on chromosome 12, and PRPF6 (rs1108981338 and rs45431912) on chromosome 17 were found in the white blood cell count QTL (SSC6: 79,653-104,254 kb; SSC12: 38,822-47,927 kb) and lymphocyte number QTL (48,731-66,928 kb) regions reported in previous studies (Reiner et al., 2008; Okamura et al., 2012). However, because there is a dearth of QTL studies related to the immune system of pigs and the QTL region is wide, high-density QTL fine mapping and additional studies should be performed to better evaluate the genes that were analysed in this study as positional candidate genes.
The authors performed functional annotation of 49 genes that were detected to be significantly associated with blood component traits using tools on the DAVID website and detected 24 GO terms in all three categories (biological process, cellular component, and molecular function). Pathway searches were performed using the GeneMANIA and KEGGParser tools and integrated interactions between candidate genes were also investigated. Only eight genes (PSMB4, PSME3, MAPKAPK3, CTLA4, CUL7, GGT1, IDH3B, and RXRB) of the 49 candidate genes were found to interact with each other, and four pathways (immune system, adaptive immune system, class I MHC-mediated antigen processing and presentation and antigen processing: ubiquitination & proteasome degradation) related to the total immune system were identified (Figure 1). In particular, the PSMB4 and PSME3 genes were actively networked with the other pathways and genes, which not only serve in cell cycle progression, proliferation, signal transduction, and degradation of abnormal proteins, and defective enzymes in metabolic regulation by the ubiquitin-proteasome pathway, but also have mechanisms involved in genetic diseases. For example, PA28 activators, which are among the two known activator types of the 26S proteasome (PA28 and PA700), are known to contain three genes, namely PSME1, PSME2 and PSME3 (Husom et al., 2004; McNaught et al., 2003; Matsushita et al., 2004). In addition, PSMB4 (proteasome subunit beta 4) is a gene encoding 20S proteasome subunit beta-7, which is known to regulate protease assembly in humans (Hirano et al., 2008; Murata et al., 2009). PSMB4 regulation is also known to be associated with carcinogenesis of various malignant types of tumour, including hepatocellular carcinomas and gliomas (Cui et al., 2006; Thaker et al., 2009). However, research on the molecular genetic function of the pig's immune system is still insufficient.
Conclusion
The results of this study suggest that the six remaining candidate genes also interact directly with the blood component traits-related immune pathways, which may be helpful in pig immunization and disease studies. Moreover, the class I MHC-mediated antigens that are expressed in all somatic cells are the most important antigens to determine the success of skin and organ transplantation in relation to immune rejection. Because the eight candidate genes that the authors found are included in the pathway related to this complex, additional studies should be performed on porcine heterologous organs. Further studies should be also conducted to provide data on the practical applications of pig production.
Acknowledgements
This research was supported by the 2016 scientific promotion programme funded by Jeju National University.
Authors' Contributions
BL and SK contributed equally to this work.
Conflict of Interest Declaration
The authors declare that they have no competing interests.
References
Barrett, J.C., Fry, B., Maller, J. & Daly, M.J., 2004. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263-265. https://doi.org/10.1093/bioinformatics/bth457 [ Links ]
Boddicker, N., Waide, E.H., Rowland, R., Lunney, J.K., Garrick, D.J., Reecy, J.M. & Dekkers, J., 2012. Evidence for a major QTL associated with host response to porcine reproductive and respiratory syndrome virus challenge. J. Anim. Sci. 90, 1733-1746. https://doi.org/10.2527/jas.2011-4464 [ Links ]
Cui, F., Wang, Y, Wang, J., Wei, K., Hu, J., Liu, F., Wang, H., Zhao, X., Zhang, X. & Yang, X., 2006. The up-regulation of proteasome subunits and lysosomal proteases in hepatocellular carcinomas of the HBx gene knockin transgenic mice. Proteomics 6, 498-504. https://doi.org/10.1002/pmic.200500218 [ Links ]
Galina-Pantoja, L., Siggens, K., Van Schriek, M. & Heuven, H., 2009. Mapping markers linked to porcine salmonellosis susceptibility. Anim. Genet. 40, 795-803. https://doi.org/10.1111/j.1365-2052.2009.01916.x [ Links ]
Hirano, Y., Kaneko, T., Okamoto, K., Bai, M., Yashiroda, H., Furuyama, K., Kato, K., Tanaka, K. & Murata, S., 2008. Dissecting β-ring assembly pathway of the mammalian 20S proteasome. EMBO J. 27, 2204-2213. https://doi.org/10.1038/emboj.2008.148 [ Links ]
Husom, A.D., Peters, E.A., Kolling, E.A., Fugere, N.A., Thompson, L.V. & Ferrington, D.A., 2004. Altered proteasome function and subunit composition in aged muscle. Arch. Biochem. Biophys. 421, 67-76. https://doi.org/10.1016/j.abb.2003.10.010 [ Links ]
Kadarmideen, H.N., von Rohr, P. & Janss, L.L., 2006. From genetical genomics to systems genetics: Potential applications in quantitative genomics and animal breeding. Mamm. Genome 17, 548-564. https://doi.org/10.1007/s00335-005-0169-x [ Links ]
Kijas, J.W., Townley, D., Dalrymple, B.P., Heaton, M.P., Maddox, J.F., McGrath, A., Wilson, P., Ingersoll, R.G., McCulloch, R. & McWilliam, S., 2009. A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PLoS One 4, e4668. https://doi.org/10.1371/journal.pone.0004668 [ Links ]
Kim, S., Li, X., Lee, Y, Choi, Y, Cho, B., Choi, B., Kim, T., Kim, J. & Kim, K.S., 2011. QTL scan for meat quality traits using high-density SNP chip analysis in cross between Korean native pig and Yorkshire. Asian-Australas. J. Anim. Sci. 24, 1184-1191. https://doi.org/10.5713/ajas.2011.11031 [ Links ]
Lee, Y.M., Alam, M., Choi, B., Kim, K.S. & Kim, J.J., 2012. A whole genome association study to detect single nucleotide polymorphisms for blood components (Immunity) in a cross between Korean native pig and Yorkshire. Asian-Australas. J. Anim. Sci. 25, 1674. https://doi.org/10.5713/ajas.2012.12503 [ Links ]
Li, X., Kim, S.W., Do, K.T., Ha, Y.K., Lee, Y.M., Yoon, S.H., Kim, H.B., Kim, J.J., Choi, B.H. & Kim, K.S., 2011. Analyses of porcine public SNPs in coding-gene regions by re-sequencing and phenotypic association studies. Mol. Biol. Rep. 38, 3805-3820. https://doi.org/10.1007/s11033-010-0496-1 [ Links ]
Lim, B., Khatun, A., Kim, S.W., Nazki, S., Jeong, C.G., Gu, S., Lee, J., Lee, K.T., Park, C.K., Lee, S.M., Kim, W.I. & Kim, K.S., 2018. Polymorphisms in the porcine CD163 associated with response to PRRSV infection. Anim. Genet. 49, 98-99. https://doi.org/10.1111/age.12630 [ Links ]
Lu, X., Liu, J.F., Gong, Y.F., Wang, Z.P., Liu, Y. & Zhang, Q., 2011. Mapping quantitative trait loci for T lymphocyte subpopulations in peripheral blood in swine. BMC Genet. 12, 79. https://doi.org/10.1186/1471-2156-12-79 [ Links ]
Lu, X., Gong, Y, Liu, J., Liu, Y. & Zhang, Q., 2014. Mapping quantitative trait loci for the lysozyme level and immunoglobulin G blocking percentage of classical swine fever virus. Genet. Mol. Res. 13, 283-290. https://doi.org/10.4238/2014.January.17.13 [ Links ]
Lunney, J.K., 2007. Advances in swine biomedical model genomics. Int. J. Biol. Sci. 3, 179. https://doi.org/10.7150/ijbs.3.179 [ Links ]
Matsushita, M., Takasaki, Y., Takeuchi, K., Yamada, H., Matsudaira, R. & Hashimoto, H., 2004. Autoimmune response to proteasome activator 28alpha in patients with connective tissue diseases. J. Rheumatol. 31, 252-259. [ Links ]
McKnite, A., Bundy, J., Moural, T., Tart, J., Johnson, T., Jobman, E., Barnes, S., Qiu, J., Peterson, D. & Harris, S., 2014. Genomic analysis of the differential response to experimental infection with porcine circovirus 2b. Anim. Genet. 45, 205-214. https://doi.org/10.1111/age.12125 [ Links ]
McNaught, K.S.P., Belizaire, R., Isacson, O., Jenner, P. & Olanow, C.W., 2003. Altered proteasomal function in sporadic Parkinson's disease. Exp. Neurol. 179, 38-46. https://doi.org/10.1006/exnr.2002.8050 [ Links ]
Murata, S., Yashiroda, H. & Tanaka, K., 2009. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 10, 104. https://doi.org/10.1038/nrm2630 [ Links ]
Okamura, T., Onodera, W., Tayama, T., Kadowaki, H., Kojima-Shibata, C., Suzuki, E., Uemoto, Y., Mikawa, S., Hayashi, T. & Awata, T., 2012. A genome-wide scan for quantitative trait loci affecting respiratory disease and immune capacity in Landrace pigs. Anim. Genet. 43, 721-729. https://doi.org/10.1111/j.1365-2052.2012.02359.x [ Links ]
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M.A., Bender, D., Maller, J., Sklar, P., De Bakker, P.I. & Daly, M.J., 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559-575. https://doi.org/10.1086/519795 [ Links ]
Reiner, G., Fischer, R., Hepp, S., Berge, T., Köhler, F. & Willems, H., 2008. Quantitative trait loci for white blood cell numbers in swine. Anim. Genet. 39, 163-168. https://doi.org/10.1111/j.1365-2052.2008.01700.x [ Links ]
Schroyen, M. & Tuggle, C.K., 2015. Current transcriptomics in pig immunity research. Mamm. Genome 26, 1-20. https://doi.org/10.1007/s00335-014-9549-4 [ Links ]
Shannon, P., Markiel, A., Ozier, O., Baliga, N.S., Wang, J.T., Ramage, D., Amin, N., Schwikowski, B. & Ideker, T., 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498-2504. https://doi.org/10.1101/gr.1239303 [ Links ]
Stephens, M., Smith, N.J. & Donnelly, P., 2001. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978-989. https://doi.org/10.1086/319501 [ Links ]
Thaker, N.G., Zhang, F., McDonald, P.R., Shun, T.Y., Lewen, M.D., Pollack, I.F. & Lazo, J.S., 2009. Identification of survival genes in human glioblastoma cells by small interfering RNA screening. Mol. Pharmacol. 76, 1246-1255. https://doi.org/10.1124/mol.109.058024 [ Links ]
Uddin, M., Clinar, M., Große-Brinkhaus, C., Tesfaye, D., Tholen, E., Juengst, H., Looft, C., Wimmers, K., Phatsara, C. & Schellander, K., 2011. Mapping quantitative trait loci for innate immune response in the pig. Int. J. Immunogenet. 38, 121-131. https://doi.org/10.1111/j.1744-313X.2010.00985.x [ Links ]
Waide, E., Tuggle, C., Seräo, N., Schroyen, M., Hess, A., Rowland, R., Lunney, J., Plastow, G. & Dekkers, J., 2017. Genomewide association of piglet responses to infection with one of two porcine reproductive and respiratory syndrome virus isolates. J. Anim. Sci. 95, 16-38. https://doi.org/10.2527/jas.2016.0874 [ Links ]
Wysocki, M., Chen, H., Steibel, J., Kuhar, D., Petry, D., Bates, J., Johnson, R., Ernst, C. & Lunney, J., 2012. Identifying putative candidate genes and pathways involved in immune response to porcine reproductive and respiratory syndrome virus (PRRSV) infection. Anim. Genet. 43, 328-332. https://doi.org/10.1111/j.1365-2052.2011.02251.x [ Links ]
Zhao, S., Zhu, M. & Chen, H., 2012. Immunogenomics for identification of disease resistance genes in pigs: a review focusing on Gram-negative bacilli. J. Anim. Sci. Technol. 3, 34. https://doi.org/10.1186/2049-1891-3-34 [ Links ]
Received 1 August 2018
Accepted 21 November 2018
First published online 5 March 2019
# Corresponding author: challengekt@jejunu.ac.kr


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}