










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Animal Science
versão On-line ISSN 2221-4062
versão impressa ISSN 0375-1589
S. Afr. j. anim. sci. vol.47 no.6 Pretoria 2017
http://dx.doi.org/10.4314/sajas.v47i6.9
Molecular cloning, sequence analysis and tissue expression of bovine imprinted ASCL2 gene
O. BamideleI, IV, #; M. De DonatoI, II; S. O. PetersIII; O. G. OmitogunIV; I. G. ImumorinI, #
IAnimal Genetics and Genomics Laboratory, International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, USA
IILaboratorio Genetica Molecular, IBB, Universidad de Oriente, Cumana, Venezuela
IIIDepartment of Animal Science, Berry College, Mount Berry, GA, USA
IVDepartment of Animal Sciences, Obafemi Awolowo University, Ile-Ife, Nigeria
ABSTRACT
Achaete-scute like-2 (ASCL2) gene is a maternally expressed gene that encodes a lineage-specific transcription factor that is essential for neurectoderm and trophectoderm development and is implicated in pre-natal and post-natal development in mammals. Using comparative genomics, various in silico sequence analyses were performed to characterize the putative imprinted ASCL2 gene. Additionally, tissue expression analysis between Angus, White Fulani, and N'Dama cattle breeds was conducted, as well as a molecular cloning of the gene transcript from bovine placenta. Four site-specific motifs were identified in the basic helix-loop-helix (bHLH) domain that are highly conserved in cattle, humans and mice. Pairwise comparisons of the coding sequence of cattle, human and mice revealed a dN/dS rate ratio that was significantly less than 1 (Z test). Two synonymous single nucleotide polymorphisms (SNPs) were found within 1 kb of the cloned complementary DNA (cDNA) in cattle. Bovine ASCL2 messenger RNA (mRNA) was relatively expressed in eight adult tissues with the ASCL2 gene differentially expressed in the muscle and skin tissues of the three cattle breeds. The conservation of these site-specific motifs for phosphorylation across the three species suggests a post-translational modification of the gene function and activity of the mammalian ASCL2 gene.
Keywords: Cattle, imprinting, motifs, phosphorylation
Introduction
Imprinted genes are important in the regulation of foetal growth, development, function of the placenta and postnatal behaviour in mammals (Reik & Dean, 2003). These are essential biological functions that are necessary for mammalian reproduction. The achaete-scute like-2 (ASCL2/Mash2) gene is the vertebrate homolog of the achaete-scute genes in Drosophila, where it is involved in neuronal determination (Johnson et al., 1990). In mammals, it has been identified as a critical gene for normal placenta development and successful pregnancies (Arnold et al., 2006). The gene is localized in human beings at the centre of the 11p15 imprinted domain, along with other genes, and has been mapped to the distal portion of mouse chromosome 7 (Lee et al., 1999; Guillemot et al., 1995). To date, the imprinting status of ASCL2 has been confirmed only in mice as there is insufficient evidence to confirm its imprinting status in humans (Guillemot et al., 1995). ASCL2 is a member of the basic helix-loop-helix (bHLH) family of transcription factors that are involved in chromosomal segregation and nervous system development in mammals (Rebhan et al., 1997). Although this gene is involved in important biological processes in most mammalian species, research has focused on its human and mouse orthologs. Imumorin et al. (2012) reported the mapping of a large cluster of imprinted genes, which included ASCL2, to the bovine chromosome BTA 29. According to that study, the imprinting region on BTA 29 comparatively maps to both the human chromosome HG 11 and mouse chromosome MM 7. Previous work on ASCL2 in cattle reported sequence homology and gene expression patterns in blastocyst, oocyte, embryo, foetal and placenta tissues (Arnold et al., 2006; Wrenzycki et al., 2001; Jiang et al., 2015). It has been shown that the levels of imprinted gene expression and gene dosage are crucial to understanding genomic imprinting (Tunster et al., 2016; Jiang et al., 2015). The objectives of this study were to perform in silico analysis of the genomic messenger RNA (mRNA), and protein sequences of ASCL2 in cattle, humans and mice, SNP detection in cloned complementary DNA (cDNA) and sub-species comparisons of the gene expression patterns in adult cattle tissues.
Materials and methods
Tissue samples were taken from two biological replicates each of three cattle breeds: Angus (Bos taurus; temperate), N'Dama (Bos taurus; tropical) and White Fulani (Bos indicus; tropical). Eight tissues (muscle, brain, liver, kidney, lymph node, spinal cord, heart and skin) of the adult Angus cattle were sampled from the slaughterhouse in the State of Pennsylvania, USA, while only the muscle and skin tissues of the White Fulani and N'Dama cattle were used in this study. They were obtained from an abattoir at Lafenwa in Abeokuta, Ogun State, Nigeria. The Angus animals were handled according to the guidelines and ethics of the Cornell University farm animal experimentation protocol for the use of animals. The sampled White Fulani, and N'Dama cattle were certified by the Department of Veterinary Services, Ministry of Agriculture, Ogun State, Nigeria.
Total RNA was purified from the sampled tissues of the adult Angus, White Fulani and N'Dama cattle breeds using the RNeasy kit (QIAGEN). The isolated RNA was treated with RNase-free DNase to remove possible contaminants. Purified total RNA (1 ug) with optical density values between 1.7 and 1.8 (260/280) and 1.9 and 2.0 (260/230) were then reverse transcribed to cDNA using the iScript™ reverse transcription supermix. The bovine ASCL2 gene was cloned from Angus placenta using the Oligo dT(20) to synthesize cDNA from total RNA (10-15 ug). The polymerase chain reaction (PCR) products were amplified by a forward (5'-CGTACCAGGGGGAGTCTTGGCAG-3') and reverse (5'-GACAGGGTGGCTGGGGTCCGAGG-3') primer set using the SMART RACE cDNA amplification kit (Clontech). The resulting PCR product was then cloned into a pGEM-T Easy Vector (Promega) and sequenced using the BigDye terminator sequencing kit, V 3.1 (Applied Biosystems).
Genomic, transcript (mRNA) and protein reference sequences (refseq) for the ASCL2 gene in the imprinted gene clusters of BTA19, HG11 and MM7 were retrieved from the GenBank database of National Centre for Biotechnology Information (NCBI) (Table 1). The 5' flanking region was defined as 2000 bp upstream refseq annotation to include the transcription start site and promoter regions of the putative imprinted genes. Computational prediction of CpG islands was performed using four web-based programs (CpG island searcher (CpGIS), CpG Promoter Detection (CpGProD), CpGREPORT and CpG-PLOT). The repetitive elements were determined using the RepeatMasker program (Smit et al., 2015). The possible transcription factor binding elements were analysed with the rVista genome browser tool. Motif analysis was performed using the MEME program (Timothy et al., 2009). The functional and specific residue patterns of the identified motif were assayed by ScanProsite and Motifscan web-based programs (Gattiker et al., 2002; Marco et al., 2007). The pair-wise data alignment option in MEGA 5.2 software (Tamura et al., 2007) was used to determine the nucleotide substitutions: number of transitions (Ts), number of transversions (Tv), and the ratio of transition to transversion (Ts/Tv) across the three species. Nucleotide diversity (Pi), number of non-synonymous substitutions (dN), synonymous substitutions (dS) and dN/dS ratio were determined from the coding sequence (CDS) using DNASP5.0 program (Librado & Rozas 2009). A phylogenetic tree was constructed using the neighbour-joining method of MEGA5.2 software (Tamura et al., 2007) by selecting the p-distance model for amino acid substitutions. A bootstrap replication (1000) test was performed to obtain the bootstrap percentage values.
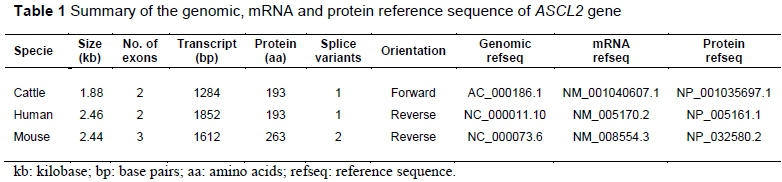
The quantitative real time polymerase chain reaction (qRT-PCR) approach used by Zaitoun & Khatib (2008) was adapted. ASCL2 gene-specific primer pair (Forward 5'-CATCCAGCCTGACCCAAGG-3' & Reverse 5'-CGTCATAAAGCCCTCTCCCC-3') was designed to amplify a 164 bp region of the mRNA sequence using the Primer 3v.0.4.0 program (Untergrasser et al., 2012). The standard guideline for designing qPCR primers effectively was adhered to. The primers were reconstituted to 10 µM concentration and optimized at 60 °C by performing a standard PCR with the cDNA as the template. The optimized gene specific primers were then used to quantitatively assay the levels of expression in the cDNA of each of the tissues. The qPCR reactions were performed following the SYBR green qPCR master mix kit protocol on a ViiA™ 7 Real-Time PCR System (Applied Biosystems, 2008). Two housekeeping genes (SDHA and GADPH) were tested to be used as reference genes across the eight tissues. GADPH (glyceraldehyde-3-phosphate dehydrogenase) was selected as the internal control gene for normalization of the fold expression because its expression was more stable. The comparative CT method for relative quantification was performed for the real time PCR analysis with liver as the calibrator (Applied Biosystems, 2008). The generated CT values were analysed using the Livak method, also known as the 2-ΔΔCT method (Livak & Schmittgen, 2001). The differential abundance of the gene expression levels for skin and muscle tissues among the three cattle breeds was analysed statistically using the independent t-test of SPSS11.0 software.
Results
CpG islands (CGI) were predicted for ASCL2 gene across cattle, human and mouse genomic refseqs. Table 2 shows the classification of the predicted CGI by the finders into promoter, intragenic and terminal CGI for each of the three species. The predicted promoter CGI identified in cattle (-1377... +1876) was found to be conserved in the promoter CGI of human (-490…-67, +157…+2718) and mouse (+352…+1885, +2211…+2293) ASCL2 gene orthologs. The analysis of the repetitive elements revealed short interspersed nuclear elements (SINE) in cattle (2.92%), humans (2.72%) and mice (6.70%) that were not of the Arthrobacter luteus (ALU) transposable repeats. Conserved transcription binding elements for E2F, NANOG and OCT4 transcription factors were found distributed across the intronic and untranslated regions (UTR). The number of binding sites was highest for E2F.
A Myc-type basic helix-loop-helix (bHLH) domain profile was identified in ASCL2 for cattle, humans and mice (Table 3). The PROSITE documentation of PDOC00038 was obtained for this domain.
It was verified in the Pfam (protein family) protein database and in the conserved domains database of NCBI with the accession number pfam00010. The location of the domain was found at the same amino acid position (50-102) and genomic region (exon 1) in both cattle and humans, which was different from mice. Five site-specific conserved residue patterns were found across the three species, with one (SKK) that was conserved only in cattle and humans. Each of the identified motifs was an active site for protein kinase cyclic adenosine monophosphate (cAMP), protein kinase C (PKC) and casein kinase C (CK2), respectively.
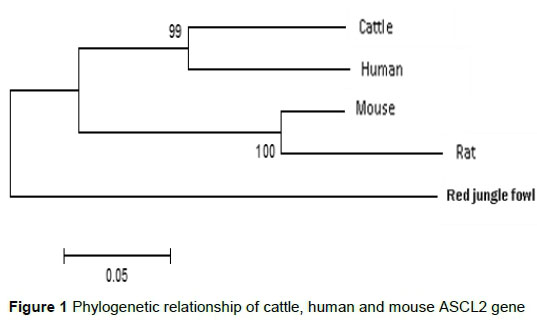
Transition bias, which is a general property of DNA sequence evolution, was observed in the cattle, human and mouse genomic sequences of ASCL2 gene. There were 257 transitions and 207 transversions as base substitution. The transition/transversion ratio (Ts/Tv : R) was 1.25, which was lower than the threshold value (2.0) of Ts/Tv. The percentage of polymorphic loci was 38.65 (657/1700), while the nucleotide diversity (Pi) was 0.21. The Pi for cattle and humans (0.23) was lowest compared with cattle/mouse (0.32) or human/mouse (0.33) pairwise comparisons. In the CDS of ASCL2 gene, the dN/dS ratio differ (Z test, P <0.05) in all the three pairwise comparisons (Table 4). Figure 1 (gene tree) showed that cattle were closely related to humans than mice in terms of their protein sequence, and common ancestor. Also, the estimates of evolutionary divergence indicated that the number of amino acid differences per site observed between cattle and humans (0.15 ± 0.03) was less than that in cattle/mouse (0.25 ± 0.03) or human/mouse (0.26 ± 0.03).
The authors obtained a 949 bp cDNA of cloned bovine ASCL2 gene in which two synonymous SNPs were identified. These were a thymine/cytosine (T/C) SNP and a thymine/adenine (T/A) SNP.
The authors analysed the relative quantification of the ASCL2 gene in all eight tissues of the Angus cattle (Table 5 and Figure 2). The results indicated that muscle, heart, and skin tissues were the most upregulated at 29.9, 80.3, and 27.9 fold, respectively, while they were downregulated in the spinal cord. The differential gene expression levels between Angus, White Fulani and N'Dama for muscle and skin tissues are represented in Figure 2. There were significant differences in the transcriptional levels of ASCL2 gene expression in both tissues across all the breeds, except for that in skin, which showed no significant difference between White Fulani and N'Dama. Among the three cattle breeds, Angus had the highest fold change of 28.95 in muscle, but the lowest fold change of 27.86 in skin, while N'Dama recorded the highest (64.00) and lowest (0.21) fold changes in skin and muscle tissues, respectively.
Discussion
DNA sequence characteristics such as CpG islands, transcription factor binding sites and repetitive elements have been used to analyse known and putative imprinted genes (De Donato et al., 2007; Khatib et al., 2007; Hutter et al., 2010). In this study, CGI were predicted for the bovine putative imprinted ASCL2 gene and in its human and mouse orthologs. Based on the prediction results, the CGI were assigned into promoter CGI, intragenic CGI and gene-terminal CGI (Bock et al., 2006). It was observed that CpGPLOT and CpGREPORT had exactly the same results because the two programs ran the same prediction criteria (Rice et al., 2000; Blankenberg 2007). The CGI regions detected by the default prediction criteria for CpGPLOT/CpGREPORT were in accordance with the method described by Gardiner-Garden & Frommer (1987). On the average, the GC content and O/E ratio of CGI predicted by CpGIS and CpGProD were the lowest in comparison with other prediction tools. This could be as a result of the stringent prediction threshold and the masking of repetitive elements, which eliminates the noise caused by sequence repeats (Ponger & Mouchiroud, 2002; Takai & Jones, 2003). From the repeat sequence analysis, the mammalian-wide interspersed repeats (MIRs) account for the SINE transposons as there were no ALUs in the ASCL2 genomic sequences of cattle, humans and mice. This was unexpected since ALUs are primate-specific SINEs (Liu et al., 2009). Imprinted loci have been reported to contain fewer SINE transposon-derived sequences than non-imprinted loci and there is a direct relationship between SINEs and imprinting (Greally, 2002).
This study identified several regions of sequence conservation in which transcription factors (TFs) were found. Although most of the TFs were present within UTR and intronic regions, the motifs may be potential sites for differential methylation (Hansen et al., 2012). Some of the regulatory elements appear to form clusters around the conserved regions found upstream of the transcription start sites with likely implications for gene silencing and regulation through their DNA binding domains. The identified TFs may need to be validated experimentally to ascertain their ability to bind to specific sites in the promoter regions, which has implications for gene expression (Tokovenko et al., 2009).
The observed transition bias indicates that during the speciation of the ASCL2 gene in cattle, humans and mice, transition base substitutions were favoured over transversions to ensure the conservation of the chemical nature of the proteins (Wakeley 1996). According to Zhao et al. (2006), base substitution is the main cause of gene variation, diversity and evolution of species. In this study, the base substitution of a cytosine (C) for thymine (T) influenced the transition bias phenomenon observed in the pairwise comparison between cattle and humans.
According to Schaffner & Sabeti (2008), natural selection is a heritable evolutionary process, which not only increases the prevalence of beneficial traits (positive selection), but also stabilizes (negative selection) traits within and across species. The stabilizing or purifying effect of negative selection is evident from a low non-synonymous amino-acid substitution rate (dN) and a dN/dS rate ratio that is less than one (Hurst 2002; Hartl & Jones 2004). It was observed in this study that the dN was lower than dS with a corresponding dN/dS rate ratio < 1 for all the three pairwise comparisons. The constraining of dN is a way by which negative selection prevents potential change to the underlying amino acids, thereby stabilizing the expression of the gene product (Wolf et al., 2009). This suggests the evolutionary stabilization of the growth-related traits of the ASCL2 gene by the selective removal of deleterious alleles across cattle, humans and mice.
The site-specific motifs identified were phosphorylation sites for each of the protein kinases. Phosphorylation is the addition of a phosphate (PO43-) group to a protein or organic molecule, thereby causing a switching off or on of that protein (Medline Database). It is an epigenetic mechanism for post-translation modification of gene function and activity (Beltrao et al., 2009). The role of protein kinases in phospho-regulation has been compared to the transcription regulatory activity of transcription factors. According to Mair (2009), just as transcription factors regulate genes by recognizing specific DNA sequences, phosphorylation also occurs at particular amino acid motifs. The specific role of each of the protein kinases in glycogen regulation (cAMP), muscle development (PKC) and cellular regulatory mechanism (CK2) within the bHLH domain could be an important molecular evolutionary source of phenotype variability across the three species (Beltrao et al., 2009). This is possible since phosphorylation modulates protein activity with a tendency to change gene expression.
The mutations identified in this study were synonymous SNPs that did not change the amino acid sequence. The novel T/A SNP identified at position 615 of the mRNA refseq has no effect on the protein sequence. The T/A SNP at mRNA and protein sequence position 726 and 151, respectively, corresponds to the reference SNP cluster identification number rs382397497 in the database of short genetic variation (Sherry et al., 2001) This SNP did not result in any change in the amino acid (Glycine) residue.
The expression of ASCL2 gene in all eight tissues supports the hypothesis that genes with promoter CGI have ubiquitous cellular functions (Antequera, 2003). Wang et al. (2014) had also reported the expression of ASCL2 in kidney, liver, heart, and muscle, although those tissues were sampled from calves. The upregulation of the ASCL2 gene in muscle, heart and skin tissues could be associated with the presence of the identified myogenic bHLH protein family, which functions primarily in the development, cell differentiation and regeneration of skeletal and cardiac myocyte cells (Cagnazzo et al., 2006). The presence of the protein kinases involved in glycogen regulation, muscle development and cellular regulation could likewise explain the observed up-regulation in muscle, heart and skin tissues.
The differential abundance of the ASCL2 gene was investigated and compared in the muscle and skin tissues of the three cattle breeds. Generally, genomic imprinting is known to be species-specific, tissue specific and developmental-stage specific, but, this study shows the presence of sub-species differences (i.e. Bos primigenius taurus and Bos primigenius indicus) in the expression profiles of ASCL2 in three cattle breeds. The authors had alluded to this sub-species specificity in a previous study (Bamidele et al., 2016). This may further our understanding of the mechanisms regulating imprinting in mammalian species.
The selection of the two tissues (muscle, skin) was based on the knowledge of their strategic economic importance in livestock production. It was observed that the Angus cattle breed had the highest differential abundance in muscle tissues among the three breeds. This suggests that the years of intensive breeding of Angus beef cattle may have increased selective pressure for growth-related genes, which may also account for its better breed performance (i.e. growth rate, feed conversion ratio) than White Fulani or N'Dama. In addition to the genetic advantage the Angus has over the White Fulani and N'Dama in terms of growth performance, nutritional factors might have contributed to the observed significant differences in the ASCL2 gene expression levels for muscle across the three breeds. The influence of nutrition on genes related to myogenesis and muscular development has been documented in several reports (Yin & Li, 2008). Assessing gene expression in the three breeds for skin tissue showed that N'Dama and White Fulani were highly abundant compared with Angus. N'Dama was found to have the highest differential abundance for ASCL2 across the breeds, though there was no significant difference between White Fulani and N'Dama. Although cattle belong to the suborder ruminantia, which is a phylogenetically distant clade to humans and rodents (Supraprimates/Euarchontoglires), comparative analyses of its proteins have been reported to be generally more similar to human orthologs than rodents (Tellam et al., 2009).
Conclusion
The in silico analyses of this study support the hypothesis that at proteomic level the bovine genome shares more homology with humans than mice. Thus, cattle is an excellent model species for human biomedical research. This work presents a contribution to sequence characterization and phylogenetic analysis of the bovine ASCL2 gene by identifying site-specific motifs associated with phospho-regulation. It is the first to report the sub-species comparisons of ASCL2 gene expression levels in cattle. The predicted promoter CGI can be used to investigate the DNA methylation status of the putative imprinted ASCL2 gene.
Acknowledgements
The authors are thankful for financial support from the College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, and Zoetis, Inc. Additional support by National Research Initiative Competitive Grant Program (Grant No. 2006-35205-16864) from the USDA National Institute of Food and Agriculture, USDA-NIFA Research Agreements (Nos. 2009-65205-05635, 2010-34444-20729) and USDA Federal Formula Hatch Funds appropriated to Cornell University Agricultural Experimental Station are gratefully acknowledged.
Authors' Contributions
IGI and OB conceptualised the idea, and designed the experiment, while OB did the laboratory work and data analyses with inputs from IGI, OGO, MDD and SOP. OB wrote the paper and SOP, OGO and IGI assisted with editing the article.
Conflict of Interest Declaration
The authors (OB, MDD, SOP, OGO and IGI) declare no conflict of interest.
References
Antequera, F., 2003. Structure, function and evolution of CpG Island promoters. Cell. Mol. Life Sci. 60(8), 1647:58. [ Links ]
Applied Biosystems, 2008. Guide to performing relative quantitation of gene expression using real-time quantitative PCR. Part number 4371095, Rev. Biol. 70p. [ Links ]
Arnold, D.R., Lefebvre, R. & Smith, L.C., 2006. Characterization of the placenta specific bovine mammalian achaete scute-like homologue 2 (Mash2) gene. Placenta 27, 1124-31. [ Links ]
Bamidele, O., Omitogun, O.G.O. & Imumorin, I.G., 2016. Tissue expression profile of 5 putative imprinted genes in bovine chromosome 29. Open J. Animal Sci. 6, 289-297. [ Links ]
Beltrao, P., Trinidad, J.C., Fiedler, D., Roguev, A., Lim, W.A., Shokat, K.M., Burlingame, A.L., & Krogan, N.J., 2009. Evolution of phosphoregulation: Comparison of phosphorylation patterns across yeast species. http://dx.doi.org/10.1371/journal.pbio.1000134 [ Links ]
Blankenberg, D., Taylor, J., Schenck, I., He, J., Zhang, Y., Ghent, M., Veerara-ghavan, N., Albert, I., Miller, W., Makova, K.D., Hardison, R.C. & Nekrutenko A.A., 2007. Framework for collaborative analysis of ENCODE data: Making large-scale analyses biologist-friendly. Genome Res. 17(6), 960-4. [ Links ]
Bock, C., Paulsen, M., Tierling, S., Mikeska, T., Lengauer, T. & Walter, J., 2006. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats and predicted DNA structure. PLoS Gen. 2(3), e26. [ Links ]
Cagnazzo, M., te Pas, M.F.W., Priem, J., de Wit, A.A.C., Pool, M.H., Davoli, R. & Russo, V., 2006. Comparison of prenatal muscle tissue expression profiles of two pig breeds differing in muscle characteristics. J. Animal Sci. 84, 1-10. [ Links ]
De Donato, M., Young, E. Z. & Imumorin I.G., 2007. Comparative analysis of the imprinted region on human chromosome 11 demonstrates differences among mammalian genomes. 15th International Conference on Plant and Animal Genome, San Diego, CA. 13-17 January 2007. [ Links ]
Gardiner-Garden M. & Frommer, M., 1987. CpG Islands in vertebrate genomes. J. Mol. Biol. 196, 261-282. [ Links ]
Gattiker, A., Gasteiger, E. & Bairoch, A., 2002. ScanProsite: A reference implementation of a prosite scanning tool. Appl. Bioinformatics 1(2),107-8. [ Links ]
Greally, J.M., 2002. Short interspersed transposable elements (SINEs) are excluded from imprinted regions in the human genome. Proc. of the Nat. Academy of Sci. 99(1), 327-332. [ Links ]
Guillemot, F., Caspary, T., Tilghman, S.M., Copeland, N.G., Gilbert, D.J., Jenkins, N.A., Anderson, D.J., Joyner, A.L., Rossant, J. & Nagy, A., 1995. Genomic imprinting of Mash2, a mouse gene required for trophoblast development. Nature Genet. 9, 235-242. [ Links ]
Hansen, K.D., Langmead, B. & Irizarry, R.A., 2012. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 13, R83.http://dx.doi.org/10.1186/gb-2012-13-10-r83 [ Links ]
Hartl, D.L. & Jones, E.W., 2004. Genetics: Analysis of genes and genomes. pp 716-759. Jones & Bartlett. http://dx.doi.org/10.1093/bioinformatics/18.4.631 [ Links ]
Hurst, L., 2002. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Gen. 18(9), 486-489. [ Links ]
Hutter, B., Bieg, M., Helms, V. & Paulsen, M., 2010. Imprinted genes show unique patterns of sequence conservation. BMC Genomics, 11, 649. http://dx.doi.org/10.1186/1471-2164-11-649. [ Links ]
Imumorin, I. G., Peters, S. O., & De Donato, M., 2012. Genomic imprinting and imprinted gene clusters in the bovine genome. In H. Khatib (ed.), Livestock Epigenetics. http://dx.doi.org/10.1002/9781119949930.ch6. [ Links ]
Jiang, Z., Dong, H., Zheng, X., Marjani, S. L., Donovan, D. M., Chen, J., & Tian, X., 2015. mRNA Levels of imprinted genes in bovine in vivo oocytes, embryos and cross species comparisons with humans, mice and pigs. Sci. Rep. 5, 17898. http://doi.org/10.1038/srep17898 [ Links ]
Johnson, J.E., Birren, S.J. & Anderson, D.J., 1990. Two rat homologues of Drosophila achaete-scute specifically expressed in neuronal precursors. Nature 346, 858-861. [ Links ]
Khatib, H., Zaitoun, I. & Kim, E.S., 2007. Comparative analysis of sequence characteristics of imprinted genes in human, mouse, and cattle. Mamm. Genome 18 (6-7), 538-547. [ Links ]
Lee, M., Brandenburg, P.S., Landes, G.M., Adams, M., Miller, G. & Feinberg, A.P., 1999. Two novel genes in the center of the 11p15 imprinted domain escape genomic imprinting. Human Mol. Genet. 8,683-690. [ Links ]
Librado, P. & Rozas, J., 2009. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451-1452. [ Links ]
Liu, G.E., Alkan, C., Jiang, L., Zhao, S. & Eichler, E.E., 2009. Comparative analysis of Alu repeats in primate genomes. Genome Res. 19(5), 876-885. [ Links ]
Livak, K.J. & Schmittgen, T.D., 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25(4), 402-408. [ Links ]
Mair, W., 2009. Change for the better: phosphoregulation of proteins drives evolution. PLoS Biol. 7(6), e1000127. doi:10.1371/journal.pbio.1000127. [ Links ]
Marco, P., Vassilios, I., Lorenzo, C., Monique, Z.C., Victor, J., Jörg, H., Olivier, M., Dmitri, K. & Laurent, F., 2007. MyHits: Improvements to an interactive re-source for analyzing protein sequences. Nucleic Acids Res. W433-W437. [ Links ]
Medline Database. http://www.nlm.nih.gov/pubs/factsheets/medline.html. Accessed 15 December 2014. [ Links ]
Ponger, L. & Mouchiroud, D., 2002. CpGProD: Identifyng CpG islands associated with transcription start sites in large genomic mammalian sequences. Bioinformatics Application Note, 18(4), 631-63. [ Links ]
Rebhan, M., Chalifa-Caspi, V., Prilusky, J. & Lancet, D., 1997. GeneCards: Integrating information about genes, proteins and diseases. Trends in Genet. 13, 163. http://dx.doi.org/10.1016/s0168-9525(97)01103-7 [ Links ]
Reik, W.F.S. & Dean, W., 2003. Mammalian epigenomics: Reprogramming the genome for development and therapy. Theriogenology 59, 21-32. [ Links ]
Rice, P., Longden, I. & Bleasby, A., 2000. EMBOSS: The European molecular biology open software suite. Trends in Genet. 16(6), 276-277. [ Links ]
Schaffner, S. & Sabeti, P., 2008. Evolutionary adaptation in the human lineage. Nature Educ. 1(1),14. [ Links ]
Sherry, S.T., Ward, M.H., Kholodov, M., Baker, J., Phan, L., Smigielski, E.M. & Sirotkin, K., 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acid Res. 29, 308-311. [ Links ]
Smit, AFA, Hubley, R & Green, P., 2015. RepeatMasker Open-4.0. http://www.repeatmasker.org [ Links ]
Takai, D. & Jones, P.A., 2003. The CpG island searcher: A new WWW re-source. In Silico Biol. 3, 235-240. [ Links ]
Tamura, K., Dudley, J., Nei, M. & Kumar, S., 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software. Mol. Biol. Evol. 24, 596-1599. [ Links ]
Tellam, R.L., Lemay, D.G., Van Tassell, C.P., Lewin, H.A., Worley, K.C. & Elsik, C.G., 2009. Unlocking the bovine genome. BMC Genomics 10, 193. http://dx.doi.org/10.1186/1471-2164-10-193 [ Links ]
Timothy, L.B., Mikael, B., Fabian, A.B., Martin, F., Charles, E.G., Luca, C., Jing-yuan, R., Wilfred, W.L. & William, S.N., 2009. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 37, W202-W208. http://dx.doi.org/10.1093/nar/gkp335 [ Links ]
Tokovenko, B., Golda, R., Protas, O., Obolenskaya, M. & El'skaya, A., 2009. COTRASIF: conservation-aided transcription factor binding site finder. Nucleic Acids Res. 37(7), e49.http://dx.doi.org/10.1093/nar/gkp084 [ Links ]
Tunster, S.J., McNamara, G.I., Creeth, H.D.J. & John R.M., 2016. Increased dosage of the imprinted ASCL2 gene restrains two key endocrine lineages of the mouse placenta. Dev. Biol. 418, 1, 55-65. https://doi.org/10.1016/j.ydbio.2016.08.014 [ Links ]
Untergrasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B.C., Remm, M. & Rozen, S.G., 2012. Primer3 - new capabilities and interfaces. Nucleic Acids Res. 40(15), e115. http://dx.doi.org/10.1093/nar/gks596 [ Links ]
Wakeley, J., 1996. The excess of transitions among nucleotide substitutions: New methods of estimating transition bias underscore its significance. Trends Ecol. Evol. 11, 158-162. http://dx.doi.org/10.1016/0169-5347 (96)10009-4 [ Links ]
Wang, M., Cui, Y., Wu, G., Li, D & Li, S., 2014. Genomic Imprinting and DNA methylation status of ASCL2 gene in cattle. Acta Veterinaria Et Zootechnica Sinica 45 (12), 1949-1956. http://dx.doi.org/10.11843/j.issn.0366-6964.2014.12.006. [ Links ]
Wolf, J.B.W., Kunstner, J., Nam, K., Jakobsson, M. & Ellegren, H., 2009. Non-linear dynamics of nonsynonymous (dN) and synonymous (dS) substitution rates affect inference of selection. Genome Biol. Evol., 1: 308-319. http://dx.doi.org/10.1093/gbe/evp030 [ Links ]
Wrenzycki, C., Wells, D., Herrmann, D., Miller, A., Oliver, J., Tervit, R. & Nie-mann, H., 2001. Nuclear transfer protocol affects messenger RNA expression pat-terns in cloned bovine blastocysts. Biol. Reprod. 65, 309e17. http://dx.doi.org/10.1095/biolreprod65.1.309 [ Links ]
Yin, J. & Li, D., 2008. Nutrigenomics approach a strategy for identification of nutrition responsive genes influencing meat edible quality traits in swine. 5th International Symposium on Recent Advances in Animal Nutrition during the 13th Animal Sciences Congress, Asian-Australasian Association of Animal Production Societies, Hanoi, Vietnam, 22-26 September 2008. [ Links ]
Zaitoun, I. & Khatib, H., 2008. Comparative genomic imprinting and expression analysis of six cattle genes. J. Anim. Sci. 86, 25-32. http://dx.doi.org/10.2527/jas.2007-0150 [ Links ]
Zhao, H., Qizhai, L., Li, J., Zeng, C., Hu, S. & Yu, J., 2006. The study of neighboring nucleotide composition and transition/transversion bias. Science in China: Series C Life Sci. 49(1), 1-8. http://dx.doi.org/10.1007/s11427-006-2002-5. [ Links ]
Received 15 June 2017
Accepted 14 August 2017
First published online 1 October 2017
# Corresponding author: bamideledeji@gmail.com, igi2@cornell.edu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}