










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Animal Science
versão On-line ISSN 2221-4062
versão impressa ISSN 0375-1589
S. Afr. j. anim. sci. vol.47 no.2 Pretoria 2017
http://dx.doi.org/10.4314/sajas.v47i2.6
ARTICLES
Modelling functional and structural impact of non-synonymous single nucleotide polymorphisms of the DQA1 gene of three Nigerian goat breeds
A. YakubuI, II, *; M. De DonatoII, III; I. G. ImumorinII, *
IDepartment of Animal Science, Nasarawa State University, Keffi, Shabu-Lafia Campus, P.M.B., 135, Lafia, 950101, Nigeria
IIAnimal Genetics and Genomics Laboratory, International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY 14853, USA
IIILaboratorio Genetica Molecular, IBB, Universidad de Oriente, Cumana, Venezuela
ABSTRACT
The DQA1 gene is a member of the highly polymorphic MHC class II locus that is responsible for the differences among individuals in immune response to infectious agents. In this study, the authors performed a comprehensive computational analysis of the functional and structural impact of non-synonymous or amino acid-changing single nucleotide polymorphisms (SNPs) (nsSNPs) that are deleterious to the DQA1 protein in Nigerian goats. A 310-bp fragment of exon 2 of the DQA1 gene was amplified and sequenced in 27 unrelated animals that are representative of three major Nigerian goat breeds (nine each of West African Dwarf, Red Sokoto, and Sahel of both sexes) using genomic DNA. Forty-two nsSNPs were identified from the alignment of the deduced amino acid sequences. Based on the PANTHER, PROVEAN and PolyPhen-2 algorithms, there was consensus in identifying the mutants I26D, E114V and V115F as being deleterious. Further, differences between the native and the mutant proteins in the subsequent molecular trajectory analysis (stabilizing and flexible residue composition, total grid energy, solvation energy, coulombic energy, solvent accessibility, and protein-protein interaction properties) revealed E114V and V115F to be highly deleterious. Combined mutational analysis comparing the amutant (I26D, E114V and V115F mutations collectively) with the native protein also showed changes that could affect protein function and structure. Further wet-lab confirmatory analysis in a pathological association study involving a larger population of goats is required at the DQA1 locus. This would lay a sound foundation for breeding disease-resistant individuals in the future.
Keywords: Goats, in silico, mutants, protein, tropics
Introduction
Single nucleotide polymorphisms (SNPs) are a common form of genetic variation among individuals (Li et al., 2009). They are amenable to high-throughput automated genotyping, and therefore are a preferred choice as genetic markers in research on diseases and their corresponding drugs (Doss et al., 2008). Non-synonymous SNPs (nsSNPs) alter the transcribed amino acid residues and result in functional diversity of the encoded proteins (Yates & Sternberg, 2013). One of the major purposes of genetics studies is to distinguish functionally neutral mutations from those that contribute to differences in phenotypes (Alshatwi et al., 2012). An abundance of diverse biological data from various sources constitutes a rich fund of knowledge, which has the power to advance our understanding of organisms. Computational methods allow the integration and effective use of these data to elucidate local and genome-wide functional connections between protein pairs, thus enabling functional inferences for uncharacterized proteins (Gray et al., 2012). Next generation sequencing techniques generate large volumes of data related to SNPs, but evaluation of biologically functional SNPs using in vitro studies is tedious, time consuming and expensive. However, the consequences of mutations and their effects on biological pathways can be predicted in silico (Patel et al., 2015; Chandramohan et al., 2015; AbdulAzeez & Borgio, 2016).
The major histocompatibility complex (MHC) is a large genomic region or gene family, which is found in most vertebrates and is highly polymorphic (Amills et al., 2005; IIhan et al., 2016). Molecules encoded by the MHC play an important role in the immune system and autoimmunity (Amills et al., 1998).
The DQA1 gene is a member of the MHC (Zhou & Hickford, 2004). Sequencing of the DQA1 complementary DNA (cDNA) revealed a single 768 bp open reading frame consisting of four exons and encoding a 255 amino acid protein (Amills et al., 2005; Plasil et al., 2016). Among MHC genes, DRB and DQA are most polymorphic (Subramani et al., 2016). This extensive structural polymorphism is responsible for the differences among individuals in immune response to infectious agents (Vandre et al., 2014). Most of the functionally important polymorphisms in DQA1 are concentrated in exon 2, and this diversity is correlated with pathogen richness (Wegner et al., 2003).
Although previous studies have examined the sequence variability of the MHC DQB1 and DRB genes (Yakubu et al., 2013; Yakubu et al., 2017), there is a dearth of information on nsSNPs of the MHC DQA1 gene of Nigerian goats. Such information could help to unravel the possible genotypes that affect livestock diseases in future association studies. Therefore, the present investigation aimed at identifying and screening deleterious nsSNPs of the DQA1 gene of West African Dwarf, Red Sokoto and Sahel goats of Nigeria, which are likely to affect the structure and function of the transcribed protein.
Materials and Methods
A 310-bp fragment of exon 2 of the MHC Class II DQA1 gene was amplified in 27 unrelated animals from three major Nigerian goat breeds (nine each of West African Dwarf (WAD), Red Sokoto (RS) and Sahel (SH)] using genomic DNA. Primers were designed using data from Amills et al. (2005). Primer sequences were DQA/FW, 5'GAAGCCCACAATGTTTGATAGTCA-3' and DQA/REV, 5'-GGGGAAGAACAACAAAGAGAGGCAG-3'. Primer-BLAST of NCBI (National Center for Biotechnology Information) was employed. The organism used was Capra hircus (taxid: 9925). The length of the forward primer was 24 bp with Tm (melting temperature) and GC (guanine-cytosine) values of 59.54 oC and 41.67%, respectively, while the corresponding length, Tm, and GC contents for the reverse primer were 25 bp, 63.73 oC and 52.00%. Primer dilution was done by adding 302.6 μL of 1 x TE buffer to DQA1 FW and 109 μL of 1 x TE buffer to DQA1 REV. Polymerase chain reaction (PCR) amplifications were carried out in a C1000TM thermal cycler (Bio-Rad, USA) in a total reaction volume of 20 μL containing about 20 ng DNA, 10 pmol of each primer in 10 μL Syd Lab PCR Premix (Syd Labs, Inc., Malden, USA) containing Taq DNA polymerase, dNTPs, MgCl2, reaction buffer, PCR stabilizer and enhancer at optimal concentrations. The thermal profile for amplifying the DQA1 exon 2 involved 35 cycles of initial denaturation at 94 °C for 4 min, denaturation at 94 °C for 30 s, annealing at 62 °C for 30 s, extension at 72 °C for 30 s, and elongation at 72 °C for 10 min. Every other protocol, including sequencing of the gene, is as described in Yakubu (2015). The ethical guidelines of the International Council for Laboratory Animal Science and Cornell University, Ithaca, NY, USA, were followed strictly. For nsSNP (missense variant) identification, sequence alignment, translations, and comparisons were carried out with the ClustalW option of MEGA 5 (Tamura et al., 2011), using the Capra_hircus_DQA1_AY464652.1 complete coding sequence (CDS) from the GenBank as the reference.
Functional effects of nsSNPs were predicted in silico using the PANTHER, PROVEAN, and PolyPhen-2 algorithms. PANTHER (Thomas et al., 2003) estimates the likelihood of a particular non-synonymous amino acid change having a functional impact on the protein. The web-based tool PROVEAN predicts the effects of an amino acid substitution or indel on the biological function of a protein, based on sequence clustering and alignment-based scoring. Variants with scores less than -2.5 were considered deleterious (Choi & Chan, 2015). PolyPhen-2 predicts the potential impact of an amino acid substitution on the function and structure of a protein (Adzhubei et al., 2010; Adzhubei et al., 2013). Where there was consensus by the three algorithms on the deleterious effect of a nsSNP, further confirmatory analyses were carried out. Additionally, a combined mutational analysis was conducted, incorporating all of the deleterious nsSNPs simultaneously (the term 'amutant' was coined to refer collectively to the nsSNPs) to reduce the possibility of false positives and exploit the effect of correlated mutations because these may enhance or diminish the functional properties of a protein.
Effects of nsSNP on protein stability were assessed with I-Mutant2.0 (Capriotti et al., 2005) and MuStab (Teng et al., 2010). I-Mutant2.0 predicts whether a single site mutation stabilizes or destabilizes the protein in 80% of the cases in which the three-dimensional structure is known and 77% of the cases in which only the protein sequence is available. MuStab, on the other hand, encodes the input sequence with the optimal feature subset, and then calls the svm_classify program of SVM-Light software package to classify the protein stability changes after the amino acid substitution (Teng et al., 2010).
Structural models for the native protein and mutant proteins I26D, E114V, V115F, and amutant were generated with Phyre2 (Kelley et al., 2015). This suite of tools aligns an input target with pre-existing templates to generate a series of predicted models. Structural similarity of alternative protein models was quantified by template modelling (Tm) scores and root mean square deviation (RMSD) in Angstroms (A) using Tm-Align. A Tm-score <0.2 is equivalent to a random structure from the PDB (Protein Data Bank) and a Tm-score of 0.5 or greater indicates that the proteins have a high probability of being in the same SCOP/CATH (structural classification of proteins/class architecture topology homology) fold (Zhang & Skolnick, 2005). An RMSD >2.0 has a negative effect on the stability and function of the protein (Han et al., 2006). Total energy after energy minimization was calculated for the native and each altered model using the Groningen Molecular Simulation (GROMOS) computer program package implementation of DeepView v 4.1 (Guex & Peitsch, 1997).
The proposed protein structures were validated with ERRAT and ProSA (Sippl, 1995; Wiederstein & Sippl, 2007). ERRAT is a program for verifying protein structures determined by crystallography (Colovos & Yeates, 1993). Error values are plotted as a function of the position of a sliding 9-residue window. The error function is based on the statistics of non-bonded atom-atom interactions in the reported structure compared with a database of reliable high-resolution structures. ProSA returns a z-score that indicates overall model quality based on the Co positions in 3-D space.
Stabilizing residues of the native and mutant DQA1 proteins of Nigerian goats were identified with SRide (Gromiha et al., 2004). The option selected was the PSSM-based encoding from the PDB file. The analysis was based on parameters such as surrounding hydrophobicity, long range order (LRO), stabilization centre and conservation score.
Residue positions that could change the conformation of the DQA1 protein were predicted with FlexPred. The software uses solvent accessibility of a protein sequence to identify the positions and thus change kinetic energy and potentially cause pathogenic disorders (Kuznetsov, 2008; Kuznetsov & McDuffie, 2008).
The molecular dynamic simulation was performed to calculate the total energy difference in solvated condition using the Poisson-Boltzmann equation (PBE) solver online tool (Smith et al., 2012; Sarkar et al., 2013). The simulation was performed by placing the protein under these conditions: i) interior dielectric constant: 4.00, ii) exterior dielectric constant: 80.00, iii) percentage fill: 80, grid scale: 2.00, iv) salt concentration: 0.10, v) probe radius: 1.40, and vi) boundary conditions: 2.00.
Solvent accessibility of residues of alternative amino acid sequences of the DQA1 protein were predicted with Weighted Ensemble Solvent Accessibility (WESA) (Shan et al., 2001; Chen & Zhou, 2005). The software predicts a residue as being buried or exposed (defined as having a surface area greater than 20% of the maximum area for that type of amino acid) with an expected accuracy of 80%.
Potential protein-protein interaction effects were predicted with InterProSurf (Zhou & Shan, 2001; Negi et al., 2006; Negi & Braun, 2007; Negi et al., 2007) and consensus Protein-Protein Interaction Site Predictor (cons-PPISP) (Chen & Zhou, 2005). InterProSurf predicts interacting amino acid residues in proteins that are most likely to interact with other proteins, given the 3D structures of subunits of a protein complex. The prediction method is based on the solvent accessible surface area of residues in the isolated subunits, a propensity scale for interface residues and a clustering algorithm to identify surface regions with residues of high interface propensities. cons-PPISP is a consensus neural network method that is trained to predict whether a surface residue is in the interaction site. Given the structure of a protein, cons-PPISP will predict the residues that would probably form the binding site for another protein.
Results
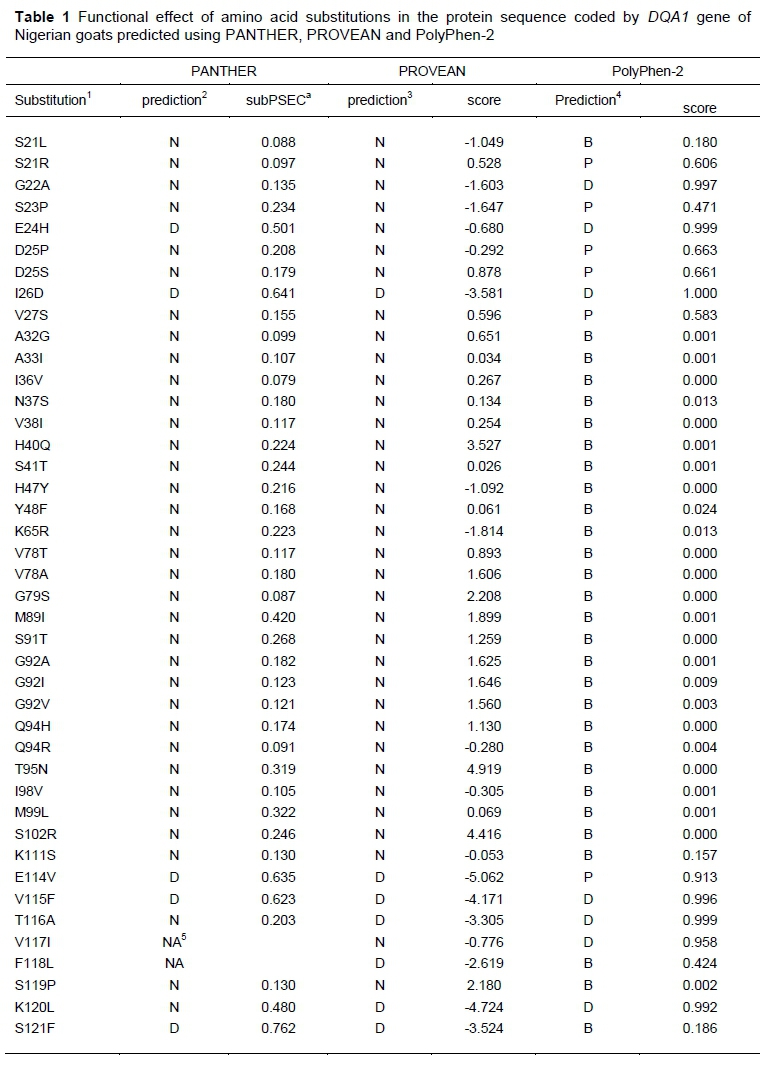
Forty-two nsSNPs of the caprine DQA1 alleles were obtained from the alignment of the deduced amino acid sequences of Nigerian goats (Table 1). Of these, five, seven and thirteen were predicted to be deleterious by PANTHER, PROVEAN and PolyPhen2, respectively (Table 1). All three algorithms identified I26D, E114V and V115F as being harmful, indicating that these amino acid substitutions negatively affect the function of the DQA1 protein. Two of the three nsSNPs that were predicted to be collectively deleterious were found to decrease protein stability, with the amino acid substitution E114V being the exception. Although the stability of E114V increased, its reliability index of 1 and 24.64% prediction confidence were very low. In contrast the reliability indexes were 8 and 9, and the prediction confidence was 94% and 88% for I26D and V115F, respectively.
There were 181 residues (71% of DQA1 sequence) modelled with 100.0% confidence by the single highest scoring template for native, I26D, E114V, V115F amino acid substitutions and the amutant protein, respectively (Figure 1). That is, the percentage identity between the DQA1 sequence of the native protein and the mutations (above) in Nigerian goats and the template used to generate the structures is high, conferring accuracy to the 3D structures. This means all the five DQA1 structures were correctly predicted.
For all of the amino acid substitutions that were deleterious, including amutant, RMSD = 0.0 and TM = 1.00. However, energy of minimization varied from one amino acid substitution to another. The values recorded for V115F and amutant (-7046.844 and -7048.283kJ/mol, respectively) were greater than for the native protein (-7208.369 kJ/mol) and I26D (-7208.369 kJ/mol) and E114V (-7209.886 kJ/mol).
The native, I26D and E114V protein structures were of higher quality (61.6%) compared with V115F and amutant (55.8%), based on the ERRAT analyses. The overall structure quality also differed between the native and the mutant proteins using ProSA [Z score = -5.57 (native), -5.57 (I26D), -5.51 (E114V), -5.54 (V115F) and -5.49 (amutant).
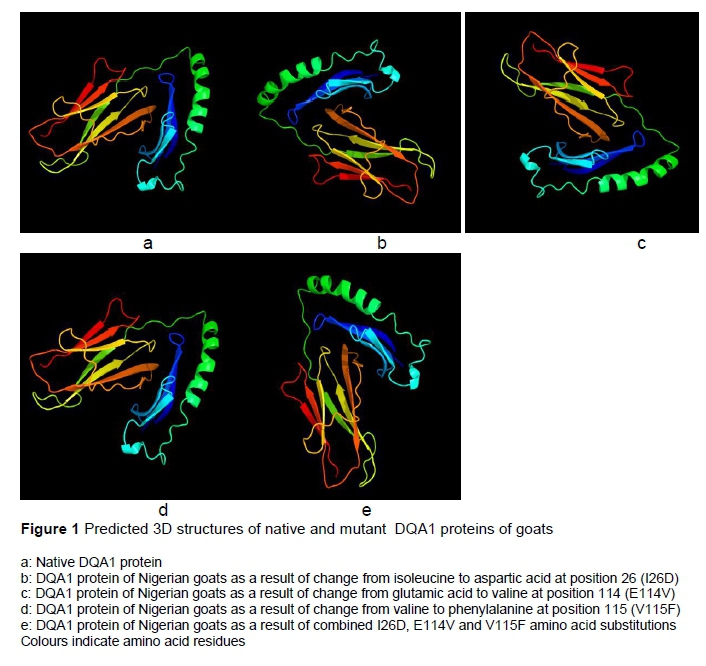
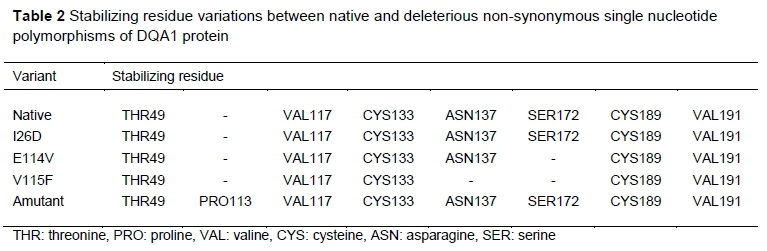
Seven stabilizing residues were predicted for the native and mutant I26D, six residues for E114V, five for V115F, and eight for amutant (Table 2). Variation in flexibility was also observed in the native and the variants. Residues 110 and 155 were unique to the native protein. The amino acid substitution E114V has a unique flexible residue at position 244, while flexible residues at positions 121 and 125 were found only in V115F (Table 3). It was not possible to calculate flexible residues for the amutant.
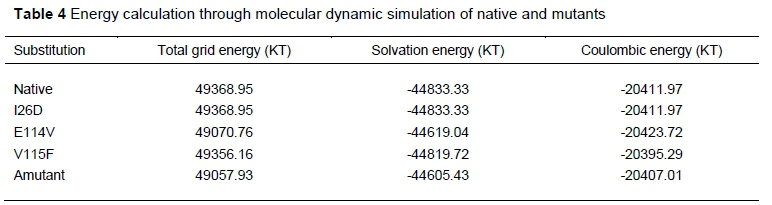
The DelPhi results showed that the native and the mutants (with the exception of I26D) differed in their average total grid energy, solvation energy and coulombic energy (Table 4).
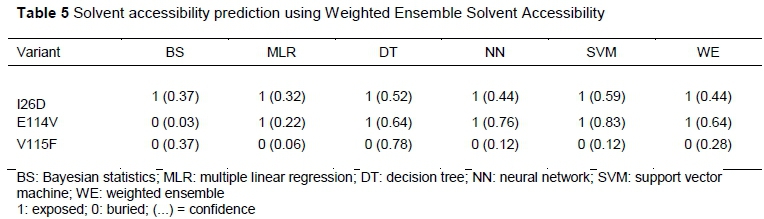
In terms of solvent accessibility, the mutant I26D was exposed, while variants E114 and V115F were found in a buried region of the protein (Table 5).
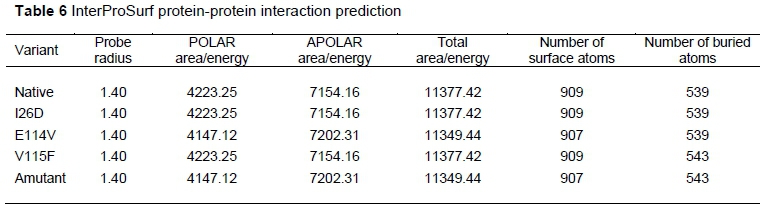
The protein-protein interaction patterns varied among the native and E114V, V115F, and amutant, respectively (Table 6). There were more buried residues in V115F and amutant (543) than in the native protein (539).
The cons-PPISP prediction of the residue contact was negative for E114V (neural network score: 0.076) and buried for V115F (Neural network score: 0.00). There was no result for the mutant I26D.
Discussion
The use of polymorphisms within genes is fast gaining attention as a complement to the current methods of selection because of their association with traits of interest in animals, especially those that affect livestock diseases (Miyasaka et al., 2011; Yakubu, 2015). However, the effects of nsSNPs in the DQA1 gene in goats have not been predicted to date in silico. Amino acid substitutions in DQA1 may induce structural and functional damage. Normal protein function can be altered by deleterious nsSNPs, through geometric constraint changes (Gromiha & Ponnuswamy, 1995), hydrophobic changes (Rose & Wolfenden, 1993), and disruption of salt bridges or hydrogen bonds (Shirley et al., 1992; Michels et al., 2011). These may have pathological phenotypic consequences (Kumar et al., 2012; Yates & Sternberg, 2014). The differences in prediction capabilities of the three algorithms used in the present study may be connected with their differing alignment procedures. The difference in the results of computational tools may be due to differences in features utilized by the methods and therefore dissimilar predictions might be expected (de Alencar & Lopes, 2010).
A structural model of a protein is important in understanding biological processes at molecular level (Wiederstein & Sippl, 2007). The statistic RMSD is commonly used to evaluate the similarity of protein structures to their templates and to determine the accuracy of the alignment of the residues of two structures (Kufareva & Abagyan, 2012). In the present study, the structures of the native and mutant proteins, which were transcribed from the DQA1 gene, appeared similar, based on RMSD values. However, energy of minimization differed between the native protein and the mutants. It implies that the higher energy of minimization of V115F and amutant might impact the protein negatively because high energy configurations may lead to physical perturbation and instability. This difference was also observed in the overall structure quality of V115F and amutant as revealed by ERRAT, where the model quality of the native protein was higher. The different energy profiles of the native and the mutants, as found in ProSA, indicate varying structural quality of the DQA1 protein of the present study. This is because the z-score of ProSA indicates overall model quality and measures the deviation of the total energy of a structure with respect to an energy distribution derived from random conformations (Sippl, 1995; Zhang & Skolnick, 1998). The more negative the z-score, the better the model quality (Saha et al., 2013). The energy separation between the native fold and the average of an ensemble of miss-folds also indicated that the conformational energy landscape of the E114V, V115F and amutant proteins differed from that of the native protein. The observed variation in the total grid energy, solvation energy, and coulombic energy of the native and the mutants could influence the structure and biological functions of the proteins. This is consistent with the submission of Smaoui et al. (2016) that mutations that increase the stability of the structure have the lowest energies, while mutations that destabilize the structure increase its energy. The varying composition of the stabilizing residues in the native and especially variants E114V, V115F and combined I26D, E114V, and V115F amino acid substitutions could influence the structure of the protein, thereby affecting its stability and function. Some of the flexible residues were found around the point of mutation in the present study. These variations may contribute to differences in folding of the protein at these positions. An inherent property of macromolecular structure is conformation flexibility (Ramesh et al., 2013). Changes in protein folding may be involved in various biological functions, such as signal transduction, catalysis, macromolecular recognition, locomotion, and many pathogenic disorders (Kuznetsov & McDuffie, 2008).
Hydrophobic core residues are probable sites of deleterious mutations (Zhangab & Wanga, 2014; David & Sternberg, 2015; Sahoo et al., 2015). Replacing a hydrophobic residue with another hydrophobic residue, as observed in mutant V115F, may induce a volume change (Eriksson et al., 1992) and thereby decrease the stability of the protein. This was supported by the submission of Ohkuri & Yamagishi (2003) that residues with larger Van der Waals volume may introduce stress in the conformation of side chains at the subunit interface. The mutant E114V is a change from a polar/hydrophilic, H-bonding, acidic residue to a non-polar/hydrophobic residue. It is possible that this change could result in loss of hydrogen bonds and, consequently, disturb correct folding (Doss & NagaSundaram, 2012). Doss et al. (2008) reported that a change from exposed to buried state could be considered functionally significant in a mutant protein. Accordingly, change in amino acid may affect polar-polar interactions within the protein molecule itself, alter the energy of stabilization, and further destabilize the protein (Peng et al., 2005). The difference in charge between hydrophobic and acidic residues, as observed in the present study, would probably disturb the ionic interaction in the native protein.
The prediction of residue solvent accessibility could assist in a better understanding of the relationship between structure and sequence (Alanazi et al., 2011). The cost of burying a hydroxyl group depends on solvent accessibility (Blaber et al., 1993). If the residue is exposed, the mutant is destabilized by <0.5 kcal/mol. If the residue is fully buried, the mutant is destabilized by ~ 1- 3 kcal/mol (Blaber et al., 1993). Mutants E114V and V115F were predicted to be buried in this study, and therefore may disturb interactions with other molecules or other parts of the protein.
Conclusion
Identified structural and functional changes in the DQA1 protein supported predictions of the nsSNPs that resulted in I26D, E114V and V115F being deleterious. The combined mutational analysis carried out with them exposed the way in which amutant is different from other individual mutations compared with the native. The difference between results of the trajectory analysis and the RMSD values may be connected with the drawback of RMSD as a positional distance-based measure compared with measurements that are contact-based. Protein-protein interaction (PPI) properties of variants E114V and V115F and amutant differed from those of the native protein. Such PPI can be strengthened or weakened by nsSNPs, which may cause loss of salt bridges, steric clashes and later post-translational modifications, among other effects. Changes in the interactions among proteins can lead to rewiring (i.e. acquisition or loss of co-complex memberships) of the PPI network and thus alter phenotype. These results could contribute to explaining susceptibility to disease in Nigerian goats, pending their confirmation in future wet lab and pathogenic population-based association studies.
Acknowledgements
This project was funded by the College of Agriculture & Life Sciences through National Research Initiative Competitive Grant Programme (Grant No. 2006-35205-16864) from the USDA National Institute of Food and Agriculture and is gratefully acknowledged. The research visit of AY to Cornell was made possible through the fellowship from the Nigerian Government Education Trust Fund (ETF), now known as Tertiary Education Trust Fund (TETFUND) (ETF/DOPS/AST&D/UNIV/KEFFI/VOL.2).
Authors' contributions
The work was designed by AY and IGI. AY, MDD and IGI did the laboratory work and statistical analysis. AY wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest statement
The authors declare that they have no conflict of interest.
References
AbdulAzeez, S. & Borgio, J.F., 2016. In-silico computing of the most deleterious nsSNPs in HBA1 gene. PLoS ONE 11, e0147702. Doi:10.1371/journal.pone.0147702. [ Links ]
Adzhubei, I., Jordan, D.M. & Sunyaev, S.R., 2013. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 7, Doi: 10.1002/0471142905.hg0720s76. [ Links ]
Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S., Sunyaev, S.R., 2010. A method and server for predicting damaging missense mutations. Nat. Methods. 7, 248-249. [ Links ]
Alanazi, M., Abduljaleel, Z., Khan, W., Warsy, A.S., Elrobh, M., Khan, Z,. Al Amri, A. & Bazzi, M.D., 2011. In silico analysis of single nucleotide polymorphism (SNPs) in human b-globin gene. PLoS ONE 6, e25876. Doi:10.1371/JOURNAL.PONE.0025876. [ Links ]
Alshatwi, A.A., Hasan, T.N., Syed, N.A., Shafi, G. & Grace, B.L., 2012. Identification of functional SNPs in BARD1 gene and in silico analysis of damaging SNPs: Based on data procured from dbSNP database. PLoS ONE 7, e43939. Doi:10.1371/journal.pone.0043939. [ Links ]
Amills, M., Ramiya, V., Nonmine, J. & Lewin, H.A., 1998. The major histocompatibility complex of ruminants. Rev. Sci. Tech. Off. Int. Epiz. 17 (1), 108-120. [ Links ]
Amills, M., Sulas, C., Sanchez, A., Bertoni, G., Zanoni, R. & Obexer-Ruff, G., 2005. Nucleotide sequence and polymorphism of the caprine major histocompatibility complex class II DQA1 (Cahi-DQA1) gene. Mol. Immunol. 42, 375-379. [ Links ]
Blaber, M., Lindstrom, J.D., Gassner, N., Xu, J., Heinz, D.W. & Matthews, B.W., 1993. Energetic cost and structural consequences of burying a hydroxyl group within the core of a protein determined from Ala-->Ser and Val-->Thr substitutions in T4 lysozyme. Biochemistry 32, 11363-11373. [ Links ]
Capriotti, E., Fariselli, P. & Casadio, R., 2005. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucl. Acids Res. 33, 306-310. [ Links ]
Chandramohan, V., Nagaraju, N., Rathod, S., Kaphle, A. & Muddapur, U., 2015. Identification of deleterious SNPs and their effects on structural level in CHRNA3 gene. Biochem. Genet. 53, 159-168. [ Links ]
Chen, H.L. & Zhou, H.X., 2005. Prediction of solvent accessibility and sites of deleterious mutations from protein sequence. Nucl. Acids Res. 33, 3193-3199. [ Links ]
Choi, Y. & Chan, A.P., 2015. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745-2747. [ Links ]
Colovos, C. & Yeates, T.O., 1993. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2 (9), 1511-15199. [ Links ]
David, A. & Sternberg, M.J.E., 2015. The contribution of missense mutations in core and rim residues of protein-protein interfaces to human disease. J. Mol. Biol. 427, 2886-2898. [ Links ]
de Alencar, S.A. & Lopes, J.C., 2010. A comprehensive in silico analysis of the functional and structural impact of SNPs in the IGF1R genel. J. Biomed. Biotechnol., 715139. Doi: 10.1155/2010/715139. [ Links ]
Doss, C.G.P. & NagaSundaram, N., 2012. Investigating the structural impacts of I64T and P311S mutations in APE1- DNA complex: A molecular dynamics approach. PLoS One 7(2), e31677. Doi: 10.1371/journal.pone.0031677 [ Links ]
Doss, C.G.P., Rajasekaran, R., Sudandiradoss, C., Ramanathan, K., Purohit, R. & Sethumadhavan R., 2008. A novel computational and structural analysis of nsSNPs in CFTR gene. Genomic Med. 2, 23-32. [ Links ]
Eriksson, A.E., Baase, W.A., Zhang, X.J., Heinz, D.W., Blaber, M., Baldwin, E.P. & Matthews, B.W., 1992. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255, 178-183. [ Links ]
Gray, V.E., Kukurba, K.R. & Kumar, S., 2012. Performance of computational tools in evaluating the functional impact of laboratory-induced amino acid mutations. Bioinformatics Discovery Note 28, 2093-2096. [ Links ]
Gromiha, M.M. & Ponnuswamy, P.K., 1995. Prediction of protein secondary structures from their hydrophobic characteristics. Int. J. Pept. Protein Res. 45, 225-240. [ Links ]
Gromiha M.M., Pujadas G., Magyar C., Selvaraj S. & Simon I. 2004. Locating the stabilizing residues in (alpha/beta)8 barrel proteins based on hydrophobicity, long-range interactions, and sequence conservation. Proteins 55, 316-329. [ Links ]
Guex, N. & Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18, 2714-2723. [ Links ]
Han, J.H., Kerrison, N., Chothia, C. & Teichmann, S.A., 2006. Divergence of interdomain geometry in two-domain proteins. Structure 14, 935-945. [ Links ]
IIhan, F., Keskin, I. & Tozluca, A., 2016. Identification of genetic variation in the major histocompatibility complex gene region in Turkish sheep breeds. S. Afr. J. Anim. Sci., 46, 366-372. [ Links ]
Kelley, L.A., Mezulis, S., Yates, C.M., Wass, M.N. & Sternberg, M.J.E., 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845-858. [ Links ]
Kufareva, I. & Abagyan R., 2012. Methods of protein structure comparison. Methods Mol. Biol. 857, 231-257. [ Links ]
Kumar, A, Rajendran, V, Sethumadhavan, R. & Purohit, R., 2012. In silico prediction of a disease-associated STIL mutant and its affect on the recruitment of centromere protein J (CENPJ). FEBS Open Bio. 2, 285-293. [ Links ]
Kuznetsov, I.B., 2008. Ordered conformational change in the protein backbone: Prediction of conformationally variable positions from sequence and low-resolution structural data. Proteins 72 (1), 74-87. [ Links ]
Kuznetsov, I.B. & McDuffie, M., 2008. FlexPred: A web-server for predicting residue positions involved in conformational switches in proteins. Bioinformation 3, 134-136. [ Links ]
Li, B., Krishnan, V.G., Mort, M.E., Xin, F., Kamati, K.K., Cooper, D.N., Mooney, S.D. & Radivojac, P., 2009. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744-2750. [ Links ]
Michels, A.W., Ostrov, D.A., Zhang, L., Nakayama, M., Fuse, M., McDaniel, K., Roep, B.O., Gottlieb, P.A., Atkinson, M.A. & Eisenbarth, G.S., 2011. Structure-based selection of small molecules to alter allele-specific MHC class II antigen presentation. J. Immunol. 187, 5921-5930. [ Links ]
Miyasaka, T., Takeshima, S.N., Matsumoto, Y., Kobayashi, N., Matsuhashi, T., Miyazaki, Y., Tanabe, Y., Ishibashi, K., Sentsui, H. & Aida, Y., 2011. The diversity of bovine MHC class II DRB3 and DQA1 alleles in different herds of Japanese Black and Holstein cattle in Japan. Gene 472 (1-2), 42-99. [ Links ]
Negi S.S. & Braun W, 2007. Statistical analysis of physical-chemical properties and prediction of protein-protein interfaces, J. Mol. Model., 13, 1157-1167 [ Links ]
Negi, S.S., Schein, C.H., Oezguen, N., Power, T.D. & Braun, W., 2007. InterProSurf: A web server for predicting interacting sites on protein surfaces. Bioinformatics 23, 3397-3399. [ Links ]
Negi S.S., Kolokoltsov A.A., Schein C.H., Davey R.A. & Braun W. 2006. Determining functionally important amino acid residues of the E1 protein of Venezuelan equine encephalitis virus, J. Mol. Model., 12, 921-929. [ Links ]
Ohkuri, T. & Yamagishi, A., 2003. Increased thermal stability against irreversible inactivation of 3-isopropylmalate dehydrogenase induced by decreased Van der Waals volume at the subunit interface. Protein Eng. 16, 215-221. [ Links ]
Patel, S.M., Koringa, P.G., Reddy, B.B., Nathani, N.M. & Joshi, C.G., 2015. In silico analysis of consequences of non- synonymous SNPs of Slc11a2 gene in Indian bovines. Genomics Data 5, 72-79. [ Links ]
Peng, L., Zhaolong, L. & John, M., 2005. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 353, 459-473. [ Links ]
Plasil, M., Mohandesan, E., Fitak, R.R., Musilova, P., Kubickova, S., Burger, P.A. & Horin, P., 2016. The major histocompatibility complex in Old World camelids and low polymorphism of its class II genes. BMC Genom. 17, 167. DOI: 10.1186/s12864-016-2500-1. [ Links ]
Ramesh, A.S., Sethumadhavan, R. & Thiagarajan, P., 2013. Structure-function studies on non-synonymous SNPs of chemokine receptor gene implicated in cardiovascular disease: A computational approach. Protein J. 32, 657-665. [ Links ]
Rose, G.D. & Wolfenden, R., 1993. Hydrogen bonding, hydrophobicity, packing, and protein folding. Annu. Rev. Biophys. Biomol. Struct. 22, 381-415. [ Links ]
Saha, C., Polash, A.H., Islam, M.T. & Shafrin, F., 2013. In silico prediction of structure and functions for some proteins of male-specific region of the human Y chromosome. Interdiscip. Sci. Comput. Life Sci. 5, 258-269. [ Links ]
Sahoo, A., Khare, S., Devanarayanan, S., Jain, P.C. & Varadarajan, R., 2015. Residue proximity information and protein model discrimination using saturation-suppressor mutagenesis. eLife. 4: e09532. [ Links ]
Sarkar, S., Witham, S., Zhang, J., Zhenirovskyy, M., Rocchia, W., Alexov, E., 2013. DelPhi Web Server: A comprehensive online suite for electrostatic calculations of biological macromolecules and their complexes. Comm. Comp. Phys. 13, 269-284. [ Links ]
Shan, Y., Wang, G., & Zhou, H.-X. 2001. Fold recognition and accurate query- template alignment by a combination of PSI-BLAST and threading. Proteins 42, 23-37. [ Links ]
Shan, Y., Wang, G. & Zhou, H.-X., 2001. Fold recognition and accurate query-template alignment by a combination of PSI-BLAST and threading. Proteins 42, 23-37. [ Links ]
Shirley, B.A., Stanssens, P., Hahn, U. & Pace, C.N., 1992. Contribution of hydrogen bonding to the conformational stability of ribonuclease T1. Biochemistry 31, 725-732 [ Links ]
Sippl, M.J. 1993. Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355-362. [ Links ]
Sippl, M.J., 1995. Knowledge-based potentials for proteins. Curr. Opin. Struct. Biol. 5, 229-235. [ Links ]
Smaoui, M.R., Mazza-Anthony, C. & Waldispühl, J., 2016. Investigating mutations to reduce Huntingtin aggregation by increasing Htt-N-Terminal stability and weakening interactions with PolyQ domain. Comput. Math. Meth. Med. http://dx.Doi.org/10.1155/2016/6247867. [ Links ]
Smith, N., Witham, S., Sarkar, S., Zhang, J., Li, L., Li, C. & Alexov, E., 2012. DelPhi Web Server v2: Incorporating atomic-style geometrical figures into the computational protocol. Bioinformatics 28, 1655-1657. [ Links ]
Subramani, K.V., Sankar, M., Raghunatha, R.R., Prasad, A. & Vikram, K., 2016. Association of genetic resistance to gastrointestinal nematodes and the polymorphism at Cahi-DQA1 exon 2. Int. J. Sci. Environ. Tech. 5, 678-687. [ Links ]
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. & Kumar, S., 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731-2739. [ Links ]
Teng, S., Srivastava, A.K. & Wang, L., 2010. Sequence feature-based prediction of protein stability changes upon amino acid substitutions. BMC Genom. 11 (Suppl 2), S5. [ Links ]
Thomas, P.D., Campbell, M.J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., Diemer, K., Muruganujan, A. & Narechania, A., 2003. Panther: A library of protein families and subfamilies indexed by function. Genome Res. 13 (9), 2129-2141. [ Links ]
Vandre, R.K., Gowane, G.R., Sharma, A.K. & Tomar, S.S., 2014. Immune responsive role of MHC class II DQA1 gene in livestock. Livest. Res. Int. 2, 1-7. [ Links ]
Wegner, K.M., Reusch, T.B.H. & Kalbe, M., 2003. Multiple parasites are driving major histocompatibility complex polymorphism in the wild. J. Evol. Biol. 16, 224-232. [ Links ]
Wiederstein, M. & Sippl, M.J. 2007. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35 (suppl 2), W407-W410. [ Links ]
Yakubu, A., 2015. Molecular diversity in major histocompatibility complex and cytokine genes and their association with heat-tolerant traits of indigenous goat breeds in Nigeria. Unpublished PhD dissertation, Department of Animal Science, University of Ibadan, Ibadan, Nigeria. 186 pp. [ Links ]
Yakubu, A., Salako, A.E., De Donato, M., Takeet, M.I., Peters, S.O., Adefenwa, M.A., Okpeku, M., Wheto, M., Agaviezor, B.O., Sanni, T.M., Ajayi, O.O., Onasanya, G.O., Ekundayo, O.J., Ilori, B.M., Amusan, S.A. & Imumorin, I.G., 2013. Genetic diversity in Exon 2 at the major histocompatibility complex DQB1 locus in Nigerian indigenous goats. Biochem. Genet. 51, 954-966. [ Links ]
Yakubu, A., Salako, A.E., De Donato, M., Peters, S.O. Takeet, M.I., Takeet, M.I., Wheto, M., Okpeku, M. & Imumorin, I.G., 2017. Association of SNP variants in MHC-Class II DRB gene with thermo-physiological indices in tropical goats. Trop. Anim. Health Prod., 49: 323-336. [ Links ]
Yates, C.M. & Sternberg, M.J., 2013. The effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein-protein interactions. J. Mol. Biol. 425, 3949-3963. [ Links ]
Zhang, L. & Skolnick, J., 1998. What should the Z-score of native protein structures be? Protein Sci. 7, 1201-1207. [ Links ]
Zhang, Y. & Skolnick, J., 2005. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic. Acids Res. 33, 2302-2309. [ Links ]
Zhangab, J. & Wanga, F., 2014. A survey and a molecular dynamics study on the (central) hydrophobic region of prion proteins. http://arxiv.org/pdf/1409.6104.pdf [ Links ]
Zhou, H. & Hickford, J.G., 2004. Allelic polymorphism in the ovine DQA1 gene. J. Anim. Sci. 82, 8-16. [ Links ]
Zhou, H.-X. & Shan, Y. 2001. Prediction of protein interaction sites from sequence profiles and residue neighbour list. Proteins 44, 336-343. [ Links ]
Received 11 December 2016
Accepted 21 January 2017
First published online 3 February 2017
* Corresponding authors: abdulmoiyak@gmail.com, igi2@cornell.edu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}