










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.113 no.3 Pretoria Mar. 2023
http://dx.doi.org/10.7196/SAMJ.2023.v113i3.16762
RESEARCH
SARS-CoV-2 mutations on diagnostic gene targets in the second wave in Zimbabwe: A retrospective genomic analysis
C NyagupeI, II; L de Oliveira MartinsIII; H GumboIV; T MasheV; T TakawiraVI; K K MaekaVII; A JuruVIII; L K ChikandaIX; A R TauyaX; A J PageXI; R A KingsleyXII; R SimbiXIII; J ChirendaXIV; J ManasaXV; V RuhanyaXVI; R T MavenyengwaXVII
IBSc, MSc; Microbiology Unit, Faculty of Medicine and Health Sciences, University of Zimbabwe, Harare, Zimbabwe
IIBSc, MSc; National Microbiology Reference Laboratory, Ministry of Health and Child Care, Harare, Zimbabwe
IIIMSc, PhD; Quadram Institute Bioscience, Norwich, UK
IVGrad Cert, MSc; National Microbiology Reference Laboratory, Ministry of Health and Child Care, Harare, Zimbabwe
VMSc, PhD; National Microbiology Reference Laboratory, Ministry of Health and Child Care, Harare, Zimbabwe
VIMSc,PhD; National Microbiology Reference Laboratory, Ministry of Health and Child Care, Harare, Zimbabwe
VIIMSc, MComPM; National Microbiology Reference Laboratory, Ministry of Health and Child Care, Harare, Zimbabwe
VIIIHBMLS, MPH; National Microbiology Reference Laboratory, Ministry of Health and Child Care, Harare, Zimbabwe
IXBSc, MSc; Microbiology Unit, Faculty of Medicine and Health Sciences, University of Zimbabwe, Harare, Zimbabwe
XBSc, MSc; Microbiology Unit, Faculty of Medicine and Health Sciences, University of Zimbabwe, Harare, Zimbabwe
XIMSc, PhD; Quadram Institute Bioscience, Norwich, UK
XIIMSc, PhD; Quadram Institute Bioscience, Norwich, UK
XIIIMSc, PhD; Directorate of Laboratory, Ministry of Health and Child Care, Harare, Zimbabwe
XIVMSc, PhD; Microbiology Unit, Faculty of Medicine and Health Sciences, University of Zimbabwe, Harare, Zimbabwe
XVMPhil, PhD; Microbiology Unit, Faculty of Medicine and Health Sciences, University of Zimbabwe, Harare, Zimbabwe
XVIMSc, PhD; Microbiology Unit, Faculty of Medicine and Health Sciences, University of Zimbabwe, Harare, Zimbabwe
XVIIMSc, PhD; Microbiology Unit, Faculty of Medicine and Health Sciences, University of Zimbabwe, Harare, Zimbabwe
ABSTRACT
BACKGROUND: SARS-CoV-2 continues to be a major issue in resource-limited settings, particularly owing to the limited supply of vaccines caused by inequitable distribution
OBJECTIVE: To monitor diagnostic gene targets to identify potential test failures caused by mutations, which is important for public health
METHODS: Here we analysed the genome sequence of SARS-CoV-2 from the second wave in Zimbabwe. A total of 377 samples were sequenced at Quadram Institute Bioscience. After quality control, 192 sequences passed and were analysed
RESULTS: The Beta variant was dominant during this period, contributing 77.6% (149) of the genomes sequenced and having a total of 2994 mutations in diagnostic polymerase chain reaction target genes. Many single nucleotide polymorphism mutations resulted in amino acid substitution that had the potential to impact viral fitness by increasing the rate of transmission or evading the immune response to previous infection or vaccination
CONCLUSION: There were nine lineages circulating in Zimbabwe during the second wave. The B.1.351 was dominant, accounting for >75%. There were over 3 000 mutations on the diagnostic genes and lineage B.1.351, contributing almost two-thirds of the mutations. The S-gene had the most mutations and the E-gene was the least mutated
The SARS-CoV-2 virus originated in Wuhan city in China in late December 2019, and was still ravaging the world a year after it was declared a pandemic by WHO in March 2020.[1] The first case in Africa was detected in Egypt in early February 2020.[2] In Zimbabwe, the first case was reported on 20 March 2020 in a returning resident coming from the UK.[3] Shortly after the first wave of infection hit Zimbabwe between April and August 2020, the second wave started around October 2020, reached its peak in the festive season, then eased in early February 2021, and was characterised by a steep rise in mortality.
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was declared a global pandemic in March 2020. In southern Africa, it was first detected on 5 and 20 March in South Africa (SA)[4] and Zimbabwe, respectively[3] In Zimbabwe, local sustained transmission was reported in April 2020, and lockdown measures were implemented. The first wave eased around August, and in October, cases started to rise again, signalling the beginning of the second wave. The virus continued to evolve, with new lineages emerging regularly. Globally, the second wave was characterised by three variants of concern (VOC), namely: the Alpha variant (Pango lineage B. 1.1.7) first identified in the UK,[5] the Beta variant (Pango lineage B.1.351) first identified in SA,[4] and the Gamma variant (Pango lineage P.1) first detected in Japan in travellers from Brazil.[1] The progression of the pandemic in Africa and onward transmission of the SARS-CoV-2 virus in different countries was associated with several variants of concern (VOC) and variants of interest (VOI), such as the Beta VOC in mid-December 2020 in SA, Pango lineage B.1.525 in Nigeria, Pango lineage A.23.1 in Uganda, B. 1.640 in the Republic of Congo and Pango lineage C.l in South Africa. There is no proven causal relationship between progression of the pandemic and development of several VOCs.[6] Many of these variants rapidly spread and replaced other circulating lineages completely, each introducing a different set of mutations, often in genes used as diagnostic targets.
SARS-CoV-2 is taxonomically classified under the order Nidovirales, family Coronaviridae, subfamily Coronavirinae. betacoronavirus genus and subgenus Sarbecovirus.[7] It is an enveloped virus with non-segmented, positive-sense, single-stranded RNA. It has lower pathogenicity than the Middle East respiratory syndrome coronavirus (MERS-CoV) that emerged in 2012, but its transmission is high. The genome of SARS-CoV-2 consists of non-segmented RNA that includes 5' and 3' untranslated regions (UTR) and several coding sequences for structural proteins, non-structural proteins and accessory proteins.[8] One of the earliest genome assemblies (Wuhan-Hu-1, accession MN908947.3) of SARS-CoV-2 is 29 903 nucleotides (nt) in length and is used as a reference to compare all subsequent mutations. It comprises a gene order of similar structure to that seen in other coronaviruses: 5'-replicase ORFlab-S-E-M-N-3', and these constitute the diagnostic targets of various reverse transcription-polymerase chain reaction (RT-PCR) kits. The ORFlab gene of Wuhan-Hu-1 is 21 291 nt in length and is cleaved into 16 non-structural proteins. Downstream open reading frames (ORFs) include the S (spike) protein, ORF3a protein, E (envelope) protein, M (membrane) protein, and N (nucleocapsid) protein, of 3 822, 828, 228, 669, and 1260 nt, respectively. The 5' and 3' terminal sequences of Wuhan-Hu-1 are also typical of Beta coronaviruses with lengths of 265 nt and 229 nt, respectively[8]
In Zimbabwe, the second wave began in October 2020 with cases rising significantly toward December 2020 and early January 2021. PCR tests were conducted at the National Microbiology Reference Laboratory (NMRL) between October 2020 and January 2021 and had a test positivity rate of 18% (3 299 of 18 000) compared with 2.2% (236 of 10 500) in the first wave between March and June 2020. Genomic surveillance revealed that the Beta variant was driving the rise in case numbers. The SARS-CoV-2 virus mutates at a rate of approximately two nucleotides per genome replication cycle,[9] which is lower than that of influenza (4 nt per genome replication cycle) or HIV (8 nt per genome replication cycle), [10] nonetheless presenting a risk that detection strategies do not accommodate this genomic change. There are many examples of this in public health diagnostics, such as a drop in the detection of Chlamydia trachomatis in Sweden,which was hailed as a public health success until genome sequencing identified a mutation that had disrupted the PCR diagnostic target and cases had increased, in Belgium a single nucleotide polymorphism (SNP) in the E gene caused a diagnostic dropout on the Roche Cobas platform,[12] and an S-gene target failure (SGTF) on the TaqPath assay following the deletion of two amino acids was used as a proxy for the Alpha variant in the UK.[5] Also in countries including Australia Denmark, Saudi Arabia, the UK and the USA, isolates containing the SNP C29200T abolished N2 detection on the Gene Xpert assay[13] Though PCR remains the gold standard for SARS-CoV-2 diagnosis, the mutations on the diagnostic genes that include ORFlab, S, N, and E may compromise the sensitivity of the kits used in its diagnosis. The test kit used during the second wave in Zimbabwe was the DaAn gene (DaAn Gene Co, Ltd, China) targeting the ORFlab and the N gene.
Genomic surveillance of SARS-CoV-2 helps to track the changes and identify mutations that may compromise the sensitivity of diagnostic targets. Moreover, it also helps to detect variants with the ability to spread more quickly, cause mild or severe disease, decreased susceptibility to therapeutics that employ monoclonal antibodies, and those with the ability to evade natural or vaccine-induced immunity. There is a paucity of data on the variants and mutations in SARS-COV-2 that circulated in the second wave in Zimbabwe. We need answers to this question to understand the evolution of the virus and its clinical and public health impact. What were the major variants and mutations and possible impacts on molecular diagnosis focusing on genes targeted in PCR assays during the second wave in Zimbabwe? In this study, we aimed to use genomic surveillance to determine mutations and variants that may have arisen or been introduced in Zimbabwe during the second wave. The objectives were to identify the variants and mutations on diagnostic genes in PCR assays used in Zimbabwe, and how they are spread in the population.
Methods
Study design and data source
We have undertaken a retrospective study of 377 routinely collected nasopharyngeal samples processed by RT-PCR at the NMRL, Harare, Zimbabwe, and sequenced at the Quadram Institute Bioscience (QIB). The samples were collected by trained clinicians in all the 10 provinces of Zimbabwe. A PCR cycle threshold (Ct) value of <30 was used to select positive samples for sequencing.
Demographic and epidemiological information was obtained, such as geographical origin, case classification obtained from the NMRLs laboratory COVID-19 request forms and the electronic laboratory information management system (LIMS), which is confidential and can only be accessed by authorised personnel through the use of a private username and password
Ethics
Ethical permission was obtained from the institution head (NMRL), the Joint Research Ethics Committee (JREC) of the University of Zimbabwe and Parirenyatwa group of hospitals (ref. no. 54/2022), and the Medical Research Council of Zimbabwe (MRCZ) (ref. no. MRCZ/B/2191). There was no specific consent obtained from the patient as the NMRL has the legal mandate to handle patient information for public health monitoring as enshrined in section 46 (notifiable diseases) of the Zimbabwe Public Health Act 2018.[14]
Specimen processing
Processing of the COVID-19 specimens was undertaken in two steps: extraction of nucleic acids, and amplification. Extraction of the nucleic acid was undertaken using the bioMériuex NucliSens Easy Mag platform, which is a semi-automated extraction machine, following the manufacturer's COVID-19 extraction protocol.[15] The extraction process is described in Box 1. Amplification of the eluates was undertaken using Quanti Studio 3, a Thermo Fisher, USA, product which is an open system.[16] The DaAn Gene RT kit for the detection of SARS-CoV-2 was used on the Quanti Studio 3 thermocycler. The DaAn Gene protocol is described in Box 1.[17]
cDNA synthesis
A total of 377 conveniently selected PCR-positive samples for SARS-CoV-2 with a Ct value <30 were selected for cDNA synthesis and were forwarded for genomic sequencing. The process of cDNA synthesis is outlined in Box 2.
Genome sequencing
The cDNA was frozen at -80°C before being packaged in dry ice and sent to QIB for sequencing using Illumina NextSeq 500 following the CoronaHiT[18] protocol. Good-quality sequences were uploaded on the Gisaid and Excel (Microsoft, USA) data of all the sequences with metadata were sent to NMRL for further mutation analysis.
Phylogenetic analysis
A phylogenetic tree was generated from all of the consensus genomes for the samples with at least 90% of the genome present using the next strain/next clade software.[19]
Data analysis
Electronically captured demographic and epidemiological data were cleaned and then imported into STATA version 12.1 (Stata Corp, USA) and Excel for analysis.
Genome analysis
Mutation rate and frequencies on the diagnostic genes were analysed by genome detective online tool version 1.137,[20]and the consensus genome was uploaded to Pangolin software[21] for lineage assignment.
Limitations of the study
The study only focused on RT-PCR SARS-CoV-2 positive samples detected by sequencing in the second wave in Zimbabwe. The conveniently selected PCR-positive samples for SARS-CoV-2 with a Ct value of <30 could have introduced some degree of bias in the study. However, some negative samples were left out for sequencing despite the individual showing clinical symptoms. False negatives may have resulted from degraded or insufficient nucleic acid, such as from excessive freeze-thaw cycles; contamination with inhibitors from the environment or DNases and RNases; poorly designed PCR; low-quality reagents; poor or inconsistent techniques during sample processing; and low-quality, faulty, or poorly calibrated equipment.[22]
Results
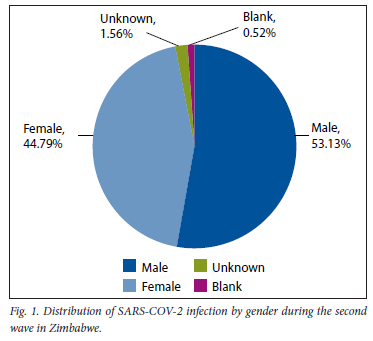
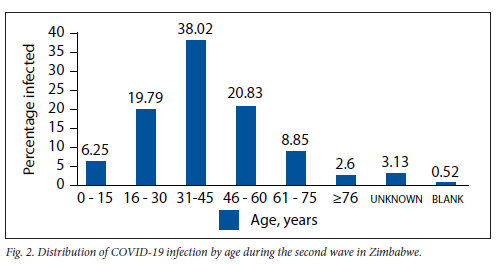
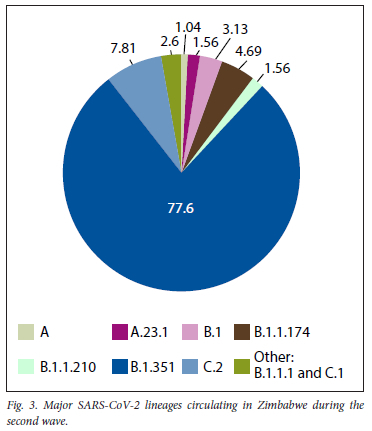
A total of 377 samples were sequenced during the second wave (October 2020 - January 2021). However, after careful selection based on quality control pass and cycle threshold value of <30, we were left with 192 samples. Of the 192 good-quality sequences, 53.13% (102) were males, 44.79% (86) were females, 1.56% (3) were of unknown gender and 0.52% (1) were designated as blank as shown in Fig. 1. The 31 - 45-year age group was the most affected, with (38.02%), followed by the 46 - 60 and 16 - 30-year age groups, with 20.83% and 19.79%, respectively, as shown in Fig. 2. The Ct value for our targets was <30. For the screening gene N, it ranged from 9.5 to 29.6, and for the ORFlab confirmatory gene, it ranged from 9.8 to 29.9 with averages of 21.7 and 23.0, respectively. The single nucleotide polymorphisms (SNP) positions on diagnostic genes amino acid positions affected and the nature or type of mutations for the sequenced samples during the second wave in Zimbabwe are shown in Table 1. The total percentage mutation and amino acid substitution of different gene targets for the variants circulating among the 192 sequenced genomes during the second wave in Zimbabwe are shown in Table 2.
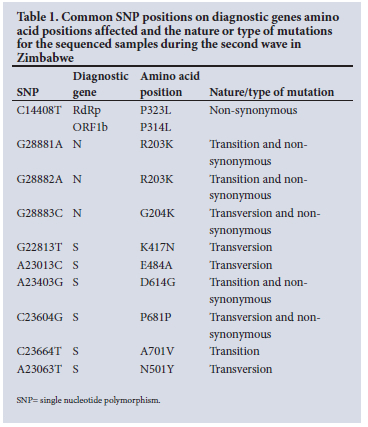
SARS-CoV-2 lineages circulating in Zimbabwe for the sequenced samples during the second wave
The VOC B. 1.351 lineage using Pangolin nomenclature, or clade 20H according to Nextstrain (https://clades.nextstrain.org) was the dominant variant circulating in Zimbabwe during the second wave, accounting for 149 (77.60%). The VOI C.2 was the second dominant variant circulating, with 15 (7.81%).[23] All the variants circulating during the second wave are shown in Fig. 3.
Phylogeny
The phylogeny tree below in Fig. 4 was constructed by loading the Zimbabwean second-wave FASTA file extracted from GISAID onto the next strain software to generate clades circulating. The 20H clade (Beta variant), or B. 1.351 lineage according to Pango, was the major circulating variant during this wave in Zimbabwe.
Results analysis
Of the 192 SARS-CoV-2 sequences, mutations resulted in a total of 3 452 nucleotide substitutions. The Beta variant lineage B.1.351, had 2 994 (86.73%) of the nucleotide substitution and was the major circulating variant during the second wave period between October 2020 and January 2021. It accounted for 77.6% of the SARS-CoV-2 variants circulating in Zimbabwe. C.2, a VOI, was the second-most common circulating, with 7.81%, followed by B. 1.174 with 4.69%. B.l, B.l. 1.210, A23.1 and A lineages were also circulating in the second wave, with the corresponding percentages of 3.13, 1.56, 1.04 and 1%, respectively.
B.1.351 lineage
The Beta variant had the most mutations on the diagnostic genes. All 149 confirmed sequences had common mutations on all the diagnostic genes. On the E-gene, all the 149 sequences had amino acid substitution on position P71L. The ORFla gene had the three most common mutations on all the 149 sequences, on positions T265I, K1655N and K3353R, while substitution at T333M and E1843D accounted for 16.67% and 16.15% of the sequences, respectively. On the ORFlb, SNP on position C14408T, which corresponds to amino acid substitution P323L on RdRP or P314L on ORFlb, was found in all the 149 sequences. SNP C14408T is poorly studied and presumably interacts with other protein-regulating catalytic activity of RdRp, resulting in considerable change in the secondary structure of position 323 or 314 in ORFlb where proline is substituted by leucine, and this was suggested to cause damage to structural integrity conferred by proline.[24] Also, viral replicative ability and transmission may potentially be affected, since RdRp participates in viral genome transcription by this SNP.[24] The S-gene is the most mutated diagnostic gene of the BÍ.351 variant accounting for 39.81% of all the mutations, with all the 149 sequences showing mutations in the following positions: D80A, D614G, and A701V. Mutations on positions D215G (93.95%), L242X (92.61%), H245X (91.95%), K417N (96.64%), E484K (60.4%) and N501Y (64.42%) were also common among the sequences. These mutations infer intrinsically higher viral fitness, particularly, high viral load A23403G (D614) and reduction in neutralising antibodies A23063T (N501Y). However, they do not impact diagnostic targets.[25] However, diagnostic impact was seen in the Alpha variant, lineage B.l. 1.7, first detected in the UK in December 2020, where an SGTF was reported in TaqPath RT-PCR kit due to 69-70 amino acid deletion.[26] This is now used as a marker of the VOC lineage B.l.1.7.[27]
C.2 lineage
This was the second-most common circulating variant in the second wave in Zimbabwe, accounting for 15 (7.81%) sequences. The 15 sequences had a total of 224 amino acid substitutions. All the diagnostic genes had one or more mutations, except for the E-gene. On the N-gene, 14 (93.33%) had non-synonymous transition SNP substitution on G28881A, G28882A (R203K) and G28883 (G204R) positions. The N-gene mutation, which includes amino acid substitution at S194L, R203K and G204R, is located in the 180247 of the N-gene, which is suggested to be a flexible linker region that lacks organised structure.[28] X-ray scattering studies suggest that this region is extended and may contain some secondary structure. S194L and G204R are conserved between MERS-CoV, SARS-CoV-1 and the reference SARS-CoV-2 genome, while R203K is conserved in SARS- CoV-1 and the reference SARS-CoV-2 genome.[28] All the 15 sequences had common mutations on position P314L of the ORFlb gene and position D614G of the S-gene. The ORFla gene showed prominent substitution on position T1246I, with 14 (93.33%) exhibiting it. The M-gene was the least mutated diagnostic target, with 2 (13.33%) of the sequences having a mutation on the M109I position.
A.23.1 lineage
This VOI, which was first isolated in Uganda, was also in circulation during the second wave in Zimbabwe, accounting for 3 (1.56%) of the circulating lineages. The isolated sequences showed no mutations on the E, M and ORFlb diagnostic genes. However, the mutation was common on the ORFla diagnostic gene with all the three sequences showing mutation on position M3752I. On the N-gene, substitution at positions S202N and N203K of two of the three sequences was witnessed. On the S-gene, a unique transition non-synonymous substitution on C23604G (P681R) was observed on two of the three sequences.
B.l lineage
This lineage, which originated in Europe, accounted for 6 (3.13%) of the variants circulating in Zimbabwe during the second wave. It shared common mutations with the SA VOC, B.1.351, on three diagnostic genes, namely, ORFla, ORFlb and S. On ORFla, substitution on position K3353R and ORFlb, the signature mutation on position P314L, is common, and the D614G on the S-gene is also common among the two lineages. On the E-gene of both B.l and B.1.351, there is a common mutation observed in positions Q71L and P71L, but a different amino acid is replaced by the same amino acid in both lineages. In B.l, the amino acid glutamine at position 71 is replaced by the amino acid leucine and in B.1.351, the amino acid proline at the same position is also replaced by the amino acid leucine.
B.l.1.1, B.l.1.74, and B.l.1.210 lineages
All three lineages have a European origin. However, they are not significant in terms of disease severity and immune evasion. They share common mutations on the diagnostic genes at position R203K on the N-gene, position P314L on the ORFlb gene and D614G on the S-gene. Mutation at position E1311A has only been shared between the B.l.1.74 and B.l.1.210 lineage, while B.l.1.1 shows no mutation on this diagnostic gene. All three lineages show no mutation in the E-gene.
A-lineage
The original Wuhan strain was also still circulating in Zimbabwe during the second wave, accounting for 2 (1.04%) of the circulating lineages. However, there were no mutations on all the diagnostic genes except the ORFla gene of one of the two sequences, which exhibited only one substitution at position N1776S, which was not present in the diagnostic genes of preceding variants.
Discussion
To the best of our knowledge, this is one of the studies that tried to analyse mutations on the diagnostic genes of SARS-CoV-2 in the sequenced samples in Zimbabwe during the second wave, and their impact on diagnostic kits, targets and immune escape. The SA VOC was the major circulating variant in Zimbabwe during the second wave, accounting for almost 80% of the circulating variants during that wave. The B.1.351 is said to have 21 plus mutations, 10 of which occur on the S-gene, which is one of the targets of the RT-PCR kit. Besides the D614G mutation, which occurs on the SI subunit of the S-gene and confers viral fitness of high viral load, the viral fitness conferred by this mutation may harbour mutagenic properties and enable the virus to spread. The A23063T (N501Y), a transversion mutation, sits on the receptor-binding domain (RBD), increasing transmission of the variant by increasing the binding of the spike protein to the angiotensin converter enzyme receptor 2 (ACE2) of the host cells, facilitating the entry of the virus in human cells.[29] SNP A23013C (E484K) and G22813T (K417N) are both transversions in nature, and are located in the RBD spike protein. The E484K mutation was found to reduce the neutralisation potency of some convalescent sera by >10-fold, and this can be a potential viral escape. SGTF was confirmed in the UK on the Thermo Fisher Taqpath RT-PCR kit with three targets ORFla, N and S-gene. There were S-gene dropouts due to amino acid 69 and 70 deletion in the Alpha VOC, lineage B.l.1.7. However, the impact on molecular diagnostic tests was found to be minimal since most of the test kits target more than one gene.[29] Similarly, all the 192 tests in this study passed quality control with good Ct values ranging from 9.4 to 29.4 for gene target 1 (ORFlab) and 10.7 to 29.9 for gene target 2 (N).
The C2 lineage had non-synonymous mutations on position R203K, and G204R of the N-gene was also detected on the latest VOC omicron, lineage B.l.1.529, detected in SA.[30] These mutations are believed to increase viral fitness survival, spread and adaptations to the human.[28] Though they are associated with a disorder in the linker region of the nucleoprotein, they have no impact on the diagnostic target of the RT-PCR kits. However, studies in countries that include Australia, Denmark, Saudi Arabia, the UK and the USA using isolates containing the SNP C29200T abolished N2 detection on the Gene Xpert assay[13] The C2 and CI lineages shared these N-gene non-synonymous mutations with the common mutation on positions C14408T(P314L) and A23403G(D614G) of the ORFlb and S-gene. However, phylogenetic analysis with international C2 lineages showed that they were interspersed, suggesting that Zimbabwe was a possible source.[23]
The A23.1 lineage, which was first identified in Uganda, had the non-synonymous mutations N202S and R203K on the N-gene and a unique P681R substitution on the S-gene, which was also found in the Delta variant in India's second wave and the new VOC B.1.1529 in SA.[31] It is located in the furin cleavage site and could increase the rate of S1-S2 cleavage, resulting in better fusogenecity and pathogenicity[31] The mutations exhibited by the sequenced samples indicated the evolution of SARS-CoV-2 into an efficient super bug regarding diagnostic target dropouts, viral fitness, spread and immune escape.
Conclusion
The Zimbabwe COVID-19 second wave was characterised by nine lineages, with the SA B. 1.351 lineage dominating the sequenced samples, constituting >75% of the circulating lineages. There were over 3 000 mutations on the diagnostic genes, with the B. 1.351 lineages contributing almost two-thirds of the mutations and the S-gene being the diagnostic gene with the most mutations. The E-gene of all the nine lineages was the least mutated diagnostic gene, with only 2/9 of the lineages possessing a common mutation P71L on B. 1.351 and B.l. A different amino acid glutamine is replaced by the same amino acid leucine. These mutations confer different attributes to the virus evolution process, ranging from a high viral load, immune evasion and spreading efficiency to antibody neutralisation. However, they had no impact on the diagnostic kit used during the second wave in Zimbabwe. Regular and efficient surveillance systems to detect circulating variants and mutations need to be put in place by the country to detect any potential harmful mutation so that public and social health measures (PSHM) are put in place to arrest the pandemic.
Declaration. This article partially fulfils the requirements of CN's MPhil degree.
Acknowledgements. The authors wish to thank Prof. R T Mavenyengwa, Dr V Ruhanya and Dr J Manasa for their unwavering support and mentorship, and Mr H Gumbo and Dr L de Oliveira Martins for sequencing support.
Author contributions. CN wrote the first draft. Funding acquisition: AJP, RAK. Leadership and supervision: AJP, RAK, RTM, VR, AG, RS, JC. Metadata curation: CN, HG, MT, TT. Samples and logistics: CN, KKM, LS. Sequencing and analysis: CN, HG, LdOM. Visualisation: CN, LdOM
Funding. The Quadram Institute authors gratefully acknowledge the support of the Biotechnology and Biological Sciences Research Council (BBSRC); their research was funded by the BBSRC Institute Strategic Programme Microbes in the Food Chain BB/R012504/1 and its constituent project BBS/E/F/000PR10352, and Quadram Institute Bioscience BBSRC funded Core Capability Grant (project number BB/CCG1860/1). For open access, the author has applied a cc-by public copyright licence to any author-accepted manuscript version arising from this submission.
Conflicts of interest. None.
References
1. European Centre for Disease, Jin Y, Yang H, et al. Virology, epidemiology, pathogenesis, and control of COVID-19. MDPI Viruses 2020;12(4):372. https://doi.org/10.3390M2040372 [ Links ]
2. Wilkinson E, Giovanetti M, Tegally H. Science 2021;374(6566):423-431. https://doi.org/10.1126/science.abj4336 [ Links ]
3. Mashe T, Takawira FT, Martins L de O, et al. Genomic epidemiology and the role of international and regional travel in the SARS-CoV-2 epidemic in Zimbabwe. A retrospective study of routinely collected surveillance data. Lancet Glob Health 2021;12.E1658-E16661. https://doi.org/10.1016/S2214-109X(21)00434-4 [ Links ]
4. Tegally H, Wilkinson E, Giovanetti M, et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. Nature 20212;592:438-443. https://doi.org/10.1038/s41586-021-03402-9 [ Links ]
5. Vofz E, Mishra S, Chand M, et al. Assessing transmissibility of SARS-CoV-2 lineage B.l. 1.7 in England. Nature 2021;593(7858):266-269. https://doi.org/10.1038/s41586-021-03470-x [ Links ]
6. European Centre for Disease Prevention and Control (ECDC). SARS-CoV-2 variants of concern as of 8 July 2021. ECDC newsletters, July 2021. [ Links ]
7. Helmy YA, Fawzy M, Elaswad A, Sobieh A, Kenney SP, Shehata AA. The COVID-19 pandemic A comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. MDPI J Clin Med 2020;9(4). https://doi.org/10.3390/jcrri9041225 [ Links ]
8. Kim J-S, Jang J-H, Kim J-M, Chung Y-S, Yoo C-K, Han M-G. Genome-wide identification and characterisation of point mutations in the SARS-CoV-2 genome. Osong Pub Health Res Perspect 2020;11(3):101-111. https://doi.org/10.24171/j.phrp.2020.11.3.05 [ Links ]
9. Page AJ, Mather AE, Viet TL, et al. Scale sequencing of SARS-CoV-2 genomes from one region allows detailed epidemiology and enables local outbreak management. Microbial Genomics 2021;7(6):000589. https://doi.org/10.1099/mgen.0.000589 [ Links ]
10. Adashek JJ, Kurzrock R Balancing clinical evidence in the context of a pandemic. Nat Biotechnol 2021;39(3):270-274.https://doi.org/10.1038/s41587-021-00834-6 [ Links ]
11. Ripa T, Nilsson P. A variant of Chlamydia trachomatis with a deletion in a cryptic plasmid. BMJ Sexual Transmitted Dis 2007;3(6):488-489. https://doi.org/10.1136/sti.2007.027698 [ Links ]
12. Artesis M, Bontems S, Gobbels P, et al. A recurrent mutation at position 26340 of SARS-CoV-2 is associated with failure of the E gene quantitative reverse. J Clin Microbiol 2020;58(10):e01598-20. https://doi.org/10.1128/JCM.01598-20 [ Links ]
13. Ziegler K, Steininger P, Ziegler R, Steinmann J, Korn K, Ensser A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Eurosurveillance 2020;25(39):2001650. https://doi.org/10.2807/1560-7917.ES.2020.25.39.2001650 [ Links ]
14. Ministry of Health and Child Care, Zimbabwe. No. 11/2018 Public Health Act Chapter 15. 171. Arrangement of Sections Sub-Part A. National health system administration. National Health Consultative Forum 2018;(II):439-514. [ Links ]
15. Sisya L. COVID-19 extraction using Biomerieux EasyMag platform SOP. Microbiology 2020,1-7. [ Links ]
16. Thermo Fisher Scientific Inc. QuantStudio 3 and 5 real-time PCR systems installation and maintenance, https://assets.iishersci.com/TFS-Assets/LSG/manuals/MAN0010407_QuantStudio3_5_InstallUseMaint_UG.pdf (accessed 7 July 2021). [ Links ]
17. Mugabe M. Laboratory NMRL 300P10 SOP. Detection kit for novel coronavirus 2019-nCoV-2 PCR fluorescent Probing DaAn Gene. 2020. [ Links ]
18. Baker DJ, Aydin A, Le-Viet T, et al. CoronaHiT. High-throughput sequencing of SARS-CoV-2 genomes. Genome Med J 2021;13(1):1-11. https://doi.org/10.1186/sl3073-021-00839-5 [ Links ]
19. Cleemput S, Dumon W, Fonseca V, et al. Genome Detective Coronavirus TypingTool for rapid identification and characterisation of novel coronavirus genomes. Bioinformatics 2020;36(11):3552-3555. https://doi.org/10.1093/bioinformatics/btaal45 [ Links ]
20. Toole AO, Scher E, Underwood A, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool.Virus Evol J 2021;7(2):veab064. https://doi.org/10.1093/ve/veab064 [ Links ]
21. Leech S. Preventing false positive and false negative PCR results. Todays Clinical Lab, 2022. https://www.clinicallab.com (accessed 20 February 2022). [ Links ]
22. Mashe T, Takawira FT, Gumbo H, et al. Surveillance of SARS-CoV-2 in Zimbabwe shows the dominance of variants of concern. Lancet Microbe 2021,2(5).el77. https://doi.org/10.1016/S2666-5247(21)00061-6 [ Links ]
23. Yuan F, Wang L, Fang Y, Wang L. Global SNP analysis of 11,183 SARS-CoV-2 strains reveals high genetic diversity. Transbound Emerg Dis 2021,68(6).3288-3304. https://doi.org/10.1111/tbed.13931 [ Links ]
24. Ruiz-Rodriguez P, Francés-Gómez C, Chiner-Omens A, López MG, Jiménez-Serrano S. Evolutionary and phenotypic characterisation of two spike mutations in European lineage 20E of SARS-CoV-2. Am Soc Microbiol 2021,12(6).e02315-21. https://doi.org/https://doi.org/10.1128/mBio.02315-21 [ Links ]
25. World Health Organization. Methods for the detection and identification of SARS-CoV-2 variants summary diagnostic screening assays of known VOCs S-gene drop out or target failure. Geneva. WHO, 2021. https://www.who.int/publications/i/item/diagnostic (accessed 14 July 2021). [ Links ]
26. Brown KA, Gubbay J, Hopkins J, et al. S-Gene target failure as a marker of variant B.l. 1.7 among SARS-CoV-2 isolates in the greater Toronto area, December 2020 to March 2021. JAMA 2021,325(20.21152116. https://doi.org/10.1001.ama.2021.5607 [ Links ]
27. Narayanan S, Ritchey JC, Patil G, et al. SARS-CoV-2 genomes from Oklahoma, United States. Front Gen J 2021,11.612571. https://doi.org/10.3389/fgene.2020.612571 [ Links ]
28. Sacks JA. Impact of novel variant on the COVID-19 diagnosis. Find Diagnostics, 2021. [ Links ]
29. World Health Organization. Brief and priority actions for the member states. Geneva. WHO, 2021. [ Links ]
30. Kumar S, Thambiraja TS, Karuppanan K, Subramaniam G. Omicron and Delta variant of SARS-CoV-2. A comparative computational study of the spike protein. J Med Virol 2021,94(4).1641-1649. https://doi.org/10.1002/jmv.27526 [ Links ]
31. Saito A, Irie T, Ruzuki R, et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2021;602(7896):300-306. https://doi.org/10.1038/s41586-021-04266-9 [ Links ]
 Correspondence:
Correspondence:
C Nyagupe
nyagupecharles@gmail.com
Accepted 10 October 2022


{kind=link}
{kind=link}
{kind=link}
{kind=link}