










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.112 no.6 Pretoria Jun. 2022
http://dx.doi.org/10.7196/SAMJ.2022.v112i6.15823
RESEARCH
Solid malignancies during the first year of life: A 20-year review at Red Cross War Memorial Children's Hospital, Cape Town, South Africa
N JauquierI; N EnglishI; A DavidsonII; S G CoxIII
IMD Department of Paediatric Surgery, Red Cross War Memorial Children's Hospital and Faculty of Health Sciences, University of Cape Town, South Africa
IIMPhil; HaematologyI Oncology Service, Red Cross War Memorial Children's Hospital and Department of Paediatrics and Child Health Faculty of Health Sciences, University of Cape Town, South Africa
IIIDCS (SA), Cert Paed Surg; Department of Paediatric Surgery, Red Cross War Memorial Children's Hospital and Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
BACKGROUND. Among paediatric tumours, two groups stand out: neonatal and infantile tumours, which respectively represent 2% and 10% of paediatric tumours. The distribution of tumours in these age groups is different from that in older children.
OBJECTIVES. Descriptive analysis of a cohort of patients treated for a solid malignancy at Red Cross War Memorial Children's Hospital (RCWMCH), Cape Town, South Africa.
METHODS. A 20-year retrospective case series review of patients aged <1 year at diagnosis was performed on data extracted from the RCWMCH oncology database.
RESULTS. Of 243 cases extracted from the database, 198 were solid tumours, of which 122 (61.1%) were included in the analysis; the 76 excluded were benign or of eye, bone or central nervous system origin and therefore did not meet the inclusion criteria. There were 38 renal malignancies (31.2%), 30 neuroblastomas (24.6%), 25 soft-tissue sarcomas (20.5%), 17 germ cell tumours/gonadal tumours (13.9%) and 12 liver tumours (9.8%). Of the patients, 119 (97.5%) had surgery, 91 (74.6%) had chemotherapy and 10 (8.2%) had radiotherapy. Tumour group 5-year survival was 78.5% for neuroblastic tumours, 79.0% for nephroblastomas, 81.5% for hepatoblastomas, 62.5% and 54.2% for rhabdomyosarcoma and non-rhabdomyosarcoma soft-tissue sarcomas, respectively, and 79.5% for malignant extracranial and extragonadal germ cell tumours. For the entire cohort, the mean follow-up was 46 months, with an estimated 5-year overall survival of 74.6%. Mortality was 21.5% and loss to follow-up 6.6%.
CONCLUSION. The distribution of tumours differs slightly from the literature, with a predominance of renal tumours over neuroblastomas. The overall mortality rate of 21.5%, the surgical complication rate of 10.9% and the 5-year overall survival of 74.6% correspond with the literature, supporting the view that a paediatric hospital in a middle-income country can achieve results similar to those in higher-income countries when international protocols are applied by a dedicated multidisciplinary team.
Paediatric tumours are rare, representing -1% of all new tumour cases each year.[1,2] Their origin, behaviour and treatment differ from adult tumours. Childhood tumours represent a spectrum of different diseases that vary in patient demographics, histological features and site of origin. In order to improve outcomes for these patients, the different age distributions of various types of tumours need to be considered in research protocols. Among paediatric tumours, two groups stand out: neonatal tumours (during the first 28 days of life) and infantile tumours (during the first year of life), which respectively represent 2% and 10% of paediatric tumours.[3,4] The distribution of paediatric tumours is different in different age groups, with neuroblastoma and nephroblastoma being prevalent in young children and osteosarcoma, for example, being more common in adolescence, and according to the literature the regional incidence of such tumours also varies.[5-14] Unlike adult tumours, which are usually recorded by primary site, childhood tumours are more meaningfully grouped by histological type and primary site based on the International Classification of Childhood Cancer (ICCC).[15]
Even if the histological type of tumour is the same, however tumours presenting in infants often differ in their behaviour and response to management compared with those in older children with the same histological diagnosis, indicating unique biological characteristics of these lesions. The diagnosis and management of tumours in the infant group present a unique set of challenges to the oncologist. These patients are particularly susceptible to the multiple complications associated with aggressive multimodal therapy[16] In addition, antineoplastic therapy may have significant long-term consequences with regard to growth and development, so dosage regimens often need to be altered.
Objectives
Red Cross War Memorial Children's Hospital (RCWMCH) is a tertiary paediatric hospital in Western Cape Province, South Africa (SA), and treats many paediatric malignancies from this province as well as further afield. The objective of this study was to do a descriptive analysis of this oncological population treated for a solid non-central nervous system malignancy at RCWMCH in terms of distribution by age category at diagnosis, gender, tumour type, management (chemotherapy, surgery and radiotherapy) including surgical complications and venous access device placement, and outcome. Benign tumours were excluded from this analysis because they are not subjected to the same aggressive multimodal and potentially harmful therapeutic regimens as malignant tumours and are managed by multiple teams within the hospital (general and plastic surgery, oncology) with multiple treatment modalities, and because many patients are lost to follow-up, making any statistical analysis non-representative.
Methods
A retrospective case series review was conducted at RCWMCH by the Division of Paediatric Surgery in collaboration with the Department of Paediatric Oncology. Data were extracted from the solid tumour database of the Department of Paediatric Oncology. The review included all children aged < 1 year at diagnosis of a solid malignancy between 1 January 1997 and 31 December 2016. Files with incomplete data were excluded from the analysis. Also excluded were children with brain, eye and bone tumours as well as haematolymphoid tumours, because they were not treated by the paediatric surgery division. Benign lesions were also excluded, except for certain subtypes of tumours that may present with more aggressive variants (e.g. mesoblastic nephroma), which were kept in the study. The data collected were age at presentation, gender, associated syndrome or genetic abnormality, histological features, anatomical site of the tumours, stage, clinical presentation, tumour markers, treatment (chemotherapy, surgery and radiotherapy), complications related to treatment, line (type, duration, complications), length of follow-up, and outcomes (remission, recurrence, death, lost to follow-up, estimated 5-year overall survival). Age categories <1 month, 1 - <6 months and 6-12 months were created based on the different resources these patients require when hospitalised. Descriptive statistics were used, and overall survival was calculated with Kaplan-Meier analysis.
Ethical considerations
The study was approved by the University of Cape Town Human Research Ethics Committee (ref. no. HREC262/2019).
Results
There were 243 cases in the database that met the inclusion criteria for age. After applying the exclusion criteria related to tumour type or incomplete data, 122 tiles (50.2%) were analysed: 25 files were excluded for incomplete data, 16 patients had haematolymphoid tumours, 4 patients were syndromic but tumour free (n=2 Beckwifh-Wiedemann, n=1 left hemihypertrophy and n=1 trisomy 21 with right nephroblastomatosis), and 50 other solid tumours (eye n=20, brain n=30, bone n=0) and 26 benign tumours were excluded (Fig. 1).

The 122 cases analysed were classified according to the 3rd edition of the ICCC (ICCC-3).[15]
Tumour distribution by age group
The median age at diagnosis was 5.5 (range 0.1 - 11.8) months. Only 23 (18.9%) of the tumours presented in the neonatal period. There were 46 cases (37.7%) diagnosed between 1 and 6 months of age and 53 (43.4%) between 6 and 12 months. Fig. 2 shows the age at presentation for each of the tumour types (neuroblastic, renal, hepatic, soft-tissue, and germ cell tumours/gonadal tumours), with an increasing incidence of neuroblastic and renal tumours as age progresses to 1 year.
Tumour distribution by gender
The distribution of tumours by gender was globally equal, with 62 boys (50.8%) and 60 girls (49.2%) (male/female ratio 1.03:1). However, when analysed according to pathology, differences in gender distribution were noted, with hepatic tumours showing a male preponderance (p=0.04) and soft-tissue sarcomas a female preponderance (p=0.05) (Fig. 3).
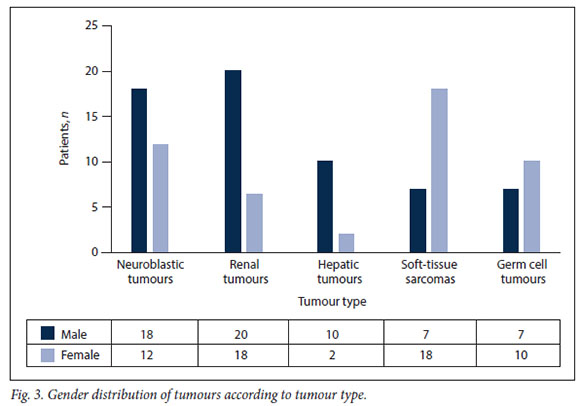
Tumour distribution in ICCC-3
Although renal tumours were the most represented in our series (n=38; 31.2%), neuroblastomas (n=30; 24.6%) were more common than Wilms' tumours (n=28; 22.9%). These were followed by soft-tissue sarcomas (n=25; 20.5%), germ cell tumours/ gonadal tumours (n=17; 13.9%), and liver tumours (n=12; 9.8%), all of which were hepatoblastomas (Table 1).

Neuroblastic tumours
Neuroblastic tumours accounted for 30 (24.6%) of the solid tumours. There were 26 neuroblastomas (21.3%) and 4 ganglio-neuroblastomas (3.3%). The median age at diagnosis was 6 (range 0-11) months. Separating neuroblastomas from ganglioneuroblastomas, the median age was 6 (0 - 11) months for the former and 10 (1 -11) months tor the latter.
Of the 30 neuroblastic tumours, 9 (30.0%) were located in the adrenal gland (5 right and 4 left), 8 (26.7%) in the mediastinum and 6 (20.0%) in the retroperitoneum (3 abdominal and 3 pelvic), and 3 (10.0%) were cervical, 2 (6.7%) thoracoabdominal, 1 (3.3%) sacrococcygeal and 1 (3.3%) sub cutaneous. Of the ganglioneuroblastomas, 2 were mediastinal, 1 cervical and 1 pelvic.
Homovanillic acid (HVA) was increased in 25 patients (83.3%), normal in 2 (6.7%) and unknown in 3 (10.0%). Specifically for ganglioneuroblastomas, HVA was normal in 1 patient and high in the other 3.
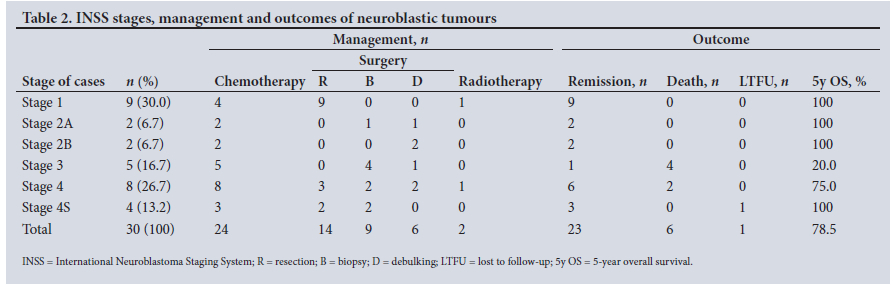
Cases were classified according to the International Neuroblastoma Staging System (INSS).[17] Table 2 shows the INSS stages and summarises the management and outcomes.
Surgery was performed on 29 children, and included 14 resections, 9 biopsies and 6 debulking procedures. Of these 29 children, 24 were treated with associated chemotherapy, 2 of whom also received radiotherapy. Those who received radiotherapy were an initially stage 1 patient who had radiotherapy for a recurrence after surgical resection, and a patient with stage 4 disease. Only one patient did not have surgery, because he died of sepsis during chemotherapy. Among the 6 children who were treated only by surgery, 5 were in stage 1 and 1 in stage 4S. Notable surgical complications, which we define as grade 2 or higher according to the Clavien-Dindo classification,'181 were 1 Horner's syndrome, 1 incisional hernia and 1 hemidiaphragmatic paralysis with perforation of the oesophagus.
The median duration of follow-up was 41 (range 0 - 142) months, and outcome analysis showed 23 remissions (76.7%), 6 deaths (20.0%) and 1 loss to follow-up (LTFU) (3.3%). The estimated 5-year overall survival for neuroblastic tumours was 78.5%.
Renal tumours
Renal tumours were the most represented in this series, with 38 cases (31.2%). They were divided into two groups: nephroblastomas (Wilms' tumours) and non-nephroblastoma renal tumours.
The median age at diagnosis was 7 (range O - 12) months. Specifically, the median age for Wilms' tumours was 7 months (2 - 12) months. For non-nephroblastoma renal tumours, the median age was 4 (0 -12) months.
Nephroblastomas
Wilms' tumours accounted for 28 (73.7%) of renal tumours and were the second most represented tumour after neuroblastic tumours.
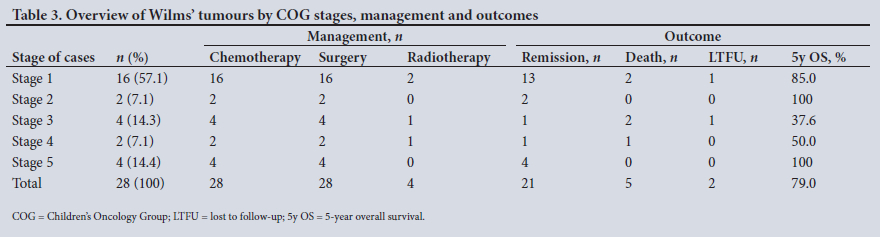
Cases were classified according to the Children's Oncology Group (COG) system.[19] Table 3 shows the COG stages and summarises management and outcomes. Among the 24 unilateral tumours, there was an equal distribution of 12 tumours of the right kidney and 12 of the left. None of the patients had a predisposing association with Wilms' tumour (Beckwith-Wiedemann, WAGR (Wilms' tumour-aniridia-genitourinary anomalies-mental retardation) or Denys-Drash syndromes), or syndromic features to suspect associated syndromes.
All the children had surgery and chemotherapy, with neoadjuvant chemotherapy given for stage 5 disease. Unilateral nephrectomy was performed in all cases except for 4 patients with stage 5 disease, in whom bilateral nephron-sparing surgery was performed. Two children in stage 1 also had radiation therapy for recurrence. It should be noted that these 2 patients had favourable histological features and there was no preoperative biopsy or capsular breach at operation. Only one stage 3 child received radiotherapy; of the other 3, 2 died rapidly and 1 was LTFU. Two children had metastatic disease (stage 4). The first had a single lung metastasis removed by thoracotomy, and the local abdominal status was stage 1. Nephrectomy was performed and he received lung radiotherapy. The second had multiple thoracic and abdominal metastases corresponding to local abdominal stage 4 disease. The tumour was unresectable and he died rapidly before further intervention.
Notable complications were 4 bowel obstructions, 2 of which required surgery, and a secondary thrombosis of the right renal artery following a left nephrectomy (histologically local stage 2, with tumour in the venous sinus), which led to right renal necrosis and the death of the child. In this patient the left renal tumour was massive, occupying the entire abdomen, and had markedly distorted the right renal vessels. The right renal artery was injured and repaired intraoperatively, with good flow, but subsequent thrombosis occurred despite anticoagulation.
The median duration of follow-up was 44 (range 0 - 132) months, and outcome analysis showed 21 remissions (75.0%), 5 deaths (17.9%) and 2 LTFU (7.1 %). The estimated 5-year overall survival for Wilms' tumour was 79.0%.
Non-nephroblastoma renal tumours
There were 10 cases in this group, divided into two groups: 6 congenital mesoblastic nephromas (CMNs) and 4 rhabdoid tumours of the kidney (RTKs) (40.0%). These tumours were found in 6 right kidneys and 4 left kidneys.
All the CMNs were localised stage 1. They were treated only surgically, without chemotherapy or radiotherapy. One child presented with an adhesive bowel obstruction requiring laparotomy 4 months after the first surgery. After a median follow-up of 63 (range 0 - 114) months, 5 patients were in remission and 1 was LTFU.
Three RTKs were initially metastatic (stage 4) and one was localised in stage 2. While the stage 2 case benefited from complete excision of the tumour, the metastatic cases were biopsied. One child with metastatic disease died soon after diagnosis, and the other 3 were treated with chemotherapy, without radiotherapy. The child in stage 2 was in remission after a 146-month follow-up, while the other 2 patients in stage 4 died from progressive local and metastatic disease at 2 and 4 months after diagnosis.
Hepatic tumours
The 12 tumours in this group represented in the series were hepatoblastomas. The median age at diagnosis was 6 (range 0 - 10) months.
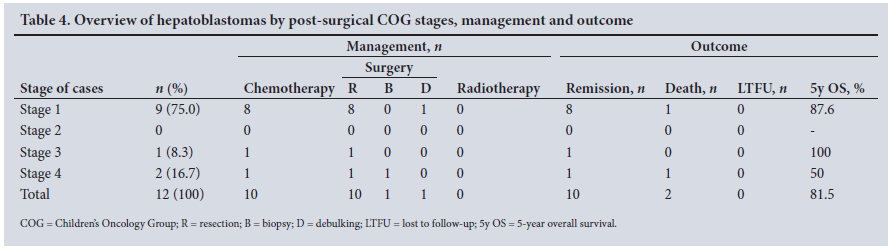
Cases were classified according to the post-surgical COG system. [20]Table 4 shows the stage, management and outcomes of patients with hepatoblastoma. Of the patients in stage 1, 2 had Beckwith-Wiedemann syndrome, one of whom also had bilateral nephroblastomatosis, omphalocele and right hemihypertrophy. He died before chemotherapy was initiated. Of the 2 patients in stage 4, one also did not have time to have chemotherapy, as he died soon after the surgical biopsy. All the patients had alpha-fetoprotein (AFP) levels well above the age standard.
With the exception of 2 children, the one with Beckwith-Wiedemann syndrome and the one with metastatic disease, who died soon after diagnosis, all other patients had neoadjuvant chemotherapy and tumour resection surgery. The patient with Beckwith-Wiedemann syndrome presented with hepatoblastoma in all liver segments, which, together with bilateral nephroblastomatosis, did not allow liver transplantation. As per protocol, no patients had radiotherapy. There were no notable surgical complications.
The median follow-up was 15 (range 0 - 149) months, and outcome analysis showed 10 remissions (83.3%) and 2 deaths (16.7%). The estimated 5-year overall survival was 81.5%.
Soft-tissue sarcomas
There were 25 patients with soft-tissue sarcomas. Of these, 7 (28.0%) were rhabdomyosarcomas (RMSs) and 18 (72.0%) were non-RMS soft-tissue sarcomas (NRSTSs). The median age at diagnosis was 4 (range 0 - 12) months. Separating RMSs and NRSTSs, the median age at presentation was 4 (0 - 12) months for RMSs and 4 (0 - 11) months for NRSTSs.
Rhabdomyosarcomas
Of the 7 RMSs, 4 (57.1%) were embryonic, 2 (28.6%) were alveolar and 1 (14.3%) was botryoid. The most frequent anatomical site was the head, with 6 cases. The other site was the bladder. None of the children had metastatic disease at presentation. Table 5 shows the pre-treatment stage, management and outcomes of patients with RMS. The TNM classification[21] was 5 (71.4%) stage 1 and 2 (28.6%) stage 3. The 2 stage 3 tumours measured >5 cm. One was located in the bladder and the other was parameningeal, both of which are unfavourable locations. All the children received neoadjuvant chemotherapy and surgery (3 biopsies, 3 excisions and 1 debulking), while only 4 received radiotherapy. There were no notable complications. The median duration of follow-up was 59 (range 5 - 170) months, and outcome analysis showed 4 remissions (57.1%), 2 deaths (28.6%) and 1 recurrence (14.3%). Regarding the causes of the 2 deaths, the patient with stage 3 disease showed progression on treatment and the patient with stage 1 disease showed progression on metronomic treatment for relapse. For the 7 patients with RMSs, the estimated 5-year overall survival was 62.5%.
Non-RMS soft-tissue sarcomas
Of the 18 NRSTSs, 8 (44.4%) were infantile fibrosacromas, 4 (22.2%) were extrarenal rhabdoid tumours, 4 (22.2%) were epithelioid haemangioendotheliomas, 1 (5.6%) was a giant cell fibroblastoma (classified intermediate risk on histology), and 1 (5.6%) was a fibrohistiocytic tumour (also intermediate histology). Table 6 shows the stage, management and outcomes of patients with NRSTSs. The TNM classification[22] was 9 (50.0%) stage 1, 5 (27.8%) stage 2, no stage 3 and 4 (22.2%) stage 4. All the patients with stage 4 disease had extrarenal rhabdoid tumours. Twelve children were treated with chemotherapy and surgery, 2 received only chemotherapy and 4 received only surgery. None of the patients had radiotherapy The notable complication in this group was section of the left facial nerve during surgical excision. The median follow-up was 32 (range 1 - 126) months, and outcome analysis showed 12 (66.7%) remissions and 6 (33.3%) deaths, including all 4 of the patients with rhabdoid tumours. For NRSTSs, the estimated 5-year overall survival was 54.2%.
Germ cell tumours and gonadal tumours
There were 17 patients with germ cell tumours and gonadal tumours, which were divided into three groups: 13 malignant extracranial and extragonadal germ cell tumours (MEEGCTs) (76.5%), 3 malignant gonadal germ cell tumours (17.6%), and 1 sex cord stromal tumour (5.9%). The median age at diagnosis was 2 (range 0 - 10) months.
Malignant extracranial and extragonadal germ cell tumours
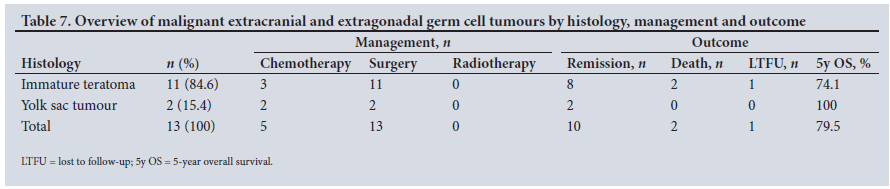
Of the 13 MEEGCTs, 11 (84.6%) were immature teratomas and 2 (15.4%) yolk sac tumours (YSTs), both of which were sacrococcygeal tumours. The immature teratomas were localised as follows: 3 in the neck, 2 sacrococcygeal, 1 on the face, 1 submental, 1 mediastinal, 1 retroperitoneal, 1 on the vulva (right lip) and 1 on the palate - this patient also had a bilateral cleft palate. The other children did not have an associated syndrome. Owing to the retrospective collection of data, staging of these tumours could not be done accurately. However, only 1 of the 13 tumours was metastatic, one of the YSTs. AFP was above the age standard in 10 children (76.9%) and within the standard in the other 3 (23.1%). Table 7 shows the histology, management and outcomes of patients with MEEGCTs. All the patients had surgical resection of the tumour and 5 had chemotherapy (n=2 YSTs and n=3 immature teratomas). The 3 patients with immature teratomas had neoadjuvant chemotherapy for three different reasons: a large neck mass with airway obstruction, an abdominal mass initially too large to resect, and patient unstable with poor overall condition. No radiotherapy was given. In terms of complications, 2 patients had bilateral vocal cord paralysis after surgery, which required tracheostomy. One of the children with vocal cord paralysis had a tumour in the neck and the other had a mediastinal tumour. The latter also had a chylothorax. The median duration of follow-up was 14 (range 0 - 105) months, and outcome analysis showed 10 remissions (76.9%), 2 deaths (15.4%) and 1 LTFU (7.7%), with an estimated 5-year overall survival of 79.5%.
Malignant gonadal germ cell tumours and sex cord stromal tumours
The 4 malignant gonadal tumours involved 2 left testicles and 2 ovaries, 1 right and 1 left. Both testicular tumours were YST stage 1[23] with increased AFP. The ovarian tumours were also stage 1,[23] and were a choriocarcinoma and a juvenile granulosa cell tumour. Tumour marker measurement were absent in both cases. None of the patients was syndromic. Table 8 shows the histology, management and outcomes of these patients. All 4 children were treated with surgery only, with no chemotherapy or radiotherapy. No notable complications were described. The 2 children with ovarian tumours were LTFU. The 2 children with testicular YSTs were in remission after 39 and 63 months of follow-up.
Lines (Fig. 4)
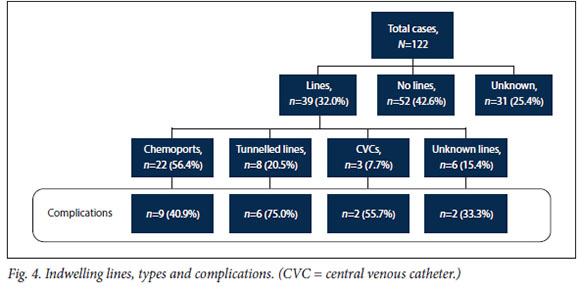
Of the 122 patients, 39 (32.0%) had surgical placement of an indwelling line for medication, 52 (42.6%) did not, and 31 (25.4%) were unknown. Of the 39 lines, 22 (56.4%) were chemoports, 8 (20.5%) tunnelled lines, 3 (7.7%) central venous catheters (CVCs) and 6 (15.4%) unknown lines. Of these lines, 19 (48.7%) had a complication that led to removal: 12 infections (63.1%) and 7 dysfunctions (36.8%). These complications involved 9 chemoports (n=6 infections and n=3 blockages), 6 tunnelled lines (n=3 infections, n=2 dislodgements and n=l fracture), 2 CVCs (n=2 infections) and 2 unknown lines (n=l infection and n=l dislodgement).
Key points of results
Overall, of the 122 cases analysed, 119 patients (97.5%) had surgery 91 (74.6%) had chemotherapy and 10 (8.2%) had radiotherapy. Of the 3 patients who did not have surgery, 1 had a cervical neuroblastoma and died of sepsis before surgery. The other 2 had epithelioid haemangioendotheliomas of the liver that were treated with chemotherapy alone. There was a total of 13 notable surgical complications, i.e. a complication rate of 10.9%. According to the Clavien-Dindo classification,[18] they are distributed as follows: 4 grade 2, 8 grade 3b and 1 grade 5. The median duration of follow-up was 46 (range 0 - 170) months, with a remission rate of 72.1% (n=88 cases), mortality of 21.5% (n=26 cases) and LTFU of 6.6% (n=8 cases). The estimated 5-year overall survival was 74.6%.
Discussion
The distribution of tumours by gender was almost equal (male/ female ratio 1.03:1), whereas when compared with data for children of all ages, there was a male predominance (1.2:1)[14, 24] However Vasilatou-Kosmidis[25] described an annual incidence of cancer in infants that was slightly higher in girls than in boys for all tumour categories. We found this female predominance only for soft-tissue sarcomas and germ cell tumours, while males presented with hepatoblastoma more frequently than females.
In terms of incidence, most publications on this age group place neuroblastoma first, followed by renal tumours, with regional differences between germ cell tumours, soft-tissue sarcomas and liver tumoursI5·9·14- 5!However, in a large series of infantile solid tumours by Jin et al[24] published in 2020, grouping 6 Beijing centres, hepatoblastomas were largely more represented than nephroblastomas. This would be explained by recruitment bias, as described by the authors.[24] On the contrary, in our series, renal tumours are in first position, followed by neuroblastic tumours, then soft-tissue sarcomas, germ cell tumours, and finally liver tumours. This finding is consistent with the results of the report by Erdmann et al.[26] on the incidence of childhood cancer in SA, except for liver tumours, which precede germ cell tumours. The position of neuroblastoma in second place behind renal tumours in terms of incidence in these results, in all paediatric age groups including children aged < 1 year, may be specific to SA. It is also interesting to note that in the study by Schickerling and Mackinnon,[27] over a 25-year period at Chris Hani Baragwanath Academic Hospital, SA, and including only children aged <3 months, the order of incidence was also different, with soft-tissue sarcoma at the forefront of malignant tumours, followed by neuroblastoma. However, if the CMNs are included, which they classified as benign tumours, the renal tumours come before the neuroblastomas, which also corresponds with our findings.
When we calculated the estimated 5-year overall survival from our results, survival rates in our cohort are comparable with the literature, in particular with the study by Alfaar et al.[7] on SEER databases (Table 9).
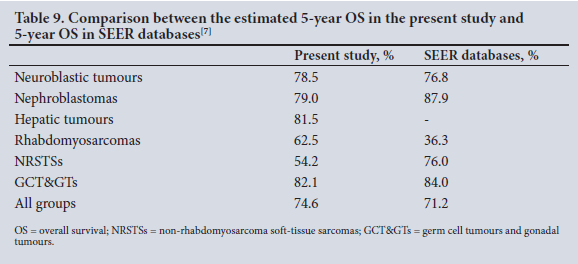
As seen in Table 9, SEER databases do not give 5-year overall survival for hepatic tumours. However, Kaatsch'141 reporteda 56% survival rate at 5 years for 0 - 14-year-olds and Jin et al.[24] a rate of 83.8%. With regard to RMSs, survival was better in our cohort compared with the SEER database, which could be explained by our small number of cases and the fact that the majority were in stage 1. However, it is interesting to note that the 5-year survival rate of 62.5% in our infant cohort corresponds with the 5-year survival rate of all children with RMS in our institution, which is 58.7% according to the study by Hendricks et al.[28] Our low 5-year survival rate for NRSTSs can be explained by the 4 cases of extrarenal rhabdoid tumour, for which the mortality rate was 100%. The prognosis of patients with rhabdoid, renal or extrarenal tumours is very poor, with only 1 of our 8 patients achieving remission. The 5-year overall survival for our entire cohort of 74.6% corresponds with the 5-year overall survival of the SEER database for patients with solid tumours.
Surgery plays a very important role in the management of solid tumours in children. Of our cohort, 97.5% had a surgical procedure in the form of resection, debulking or a biopsy, in accordance with the guidelines for the management of solid tumours in children. Whether for neuroblastic tumours,[29] renal tumours,[19] liver tumours,[30] soft-tissue tumours[31] or germ cell tumours and gonadal tumours,[23] surgery has a well-established place in the management of these children.
A surgical complication rate of 10.9% corresponds with the literature. The review by Warmann et al[32] describes rates ranging from 8% to 40%, depending on the type of tumour. Our rate of 10.9% is possibly a little underestimated, as grade 1 complications (defined as any deviation from the normal postoperative course without the need for pharmacological treatment or surgical, endoscopic and radiological interventions) are not always traceable in medical records. However, in view of the benign nature of these grade 1 complications, it can be assumed that their impact on the child's further treatment is not significant.
All 10 radiotherapies were performed according to the respective management protocols for each tumour type, i.e. 2 neuroblastomas, 4 nephroblastomas and 4 RMSs. Specifically, the need for radiotherapy in 1 patient with stage 1 neuroblastoma was due to tumour relapse.
Concerning the central lines, analysis of the results is limited because information was missing or unreliable in a quarter of the cases. However, it can be observed that while the proportion of patients with a percutaneous central line is low (32.0%), the complication rate of almost 50% suggests that more formal long-term lines should be placed. With complications affecting 75.0% of tunnelled lines, it would also appear that these are inappropriate in such young oncology patients. Our complication rate for chemoports, while being the lowest of all types of lines at 40.9%, is still far too high when compared with the systematic review by Ullman et αΙ.,[Ώwhich concluded that the complication rate in oncology patients was 15.2% for chemoports and 31.1% for tunnelled lines. Our high rates of complications have led to more appropriate device procurement, institution of bundles of care related to line sepsis, and more frequent use of chemoports as opposed to externally exposed lines.
Study limitations
This descriptive analysis has the limitation expected of a retrospective study, i.e. the 10% of files with incomplete data that excluded them de facto from the analysis. In addition, a description of the type of chemotherapy used in each case was not possible owing to the heterogeneity of the data, nor was a more precise description of radiotherapy, although the children were treated according to the COG or International Society of Paediatric Oncology protocols for each situation.
Conclusion
Neonatal and infantile tumours are rare, so the literature describing these cases is not very extensive. This descriptive analysis of a relatively large number of cases from a single centre provides additional data to integrate with the existing information. It is important to note that a paediatric hospital located in a middle-income country can achieve results similar to those in higher-income countries when international protocols are applied by a dedicated multidisciplinary team.
Declaration. None.
Acknowledgements. None.
Author contributions. NJ, SC, AD: research concept; NJ, SC: protocol writing and ethics application; NJ: data analysis and manuscript redaction; NE: data collection; NJ, AD and SC: manuscript writing and revision.
Funding. None.
Conflicts of interest. None.
References
1. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence andmortality rates andtrends- an update. Cancer Epidemiol Biomarkers Prev 201425(1). 16-27. https://doi.org/10.1158/1055-9965.EPI-15-0578 [ Links ]
2. Steliarova-Foucher E, Colombet M, Ries LAG, et al. International incidence of childhood cancer, 2001 -10! A population-based registry study. Lancet Oncol 2017; 18(6).719-731. https://doi.org/10.1016/S1470-2045(17)30186-9 [ Links ]
3. Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, eds. Cancer Incidence and Survival among Children and Adolescents. United States SEER Program 1975 - 1995. Bethesda, Md.! National Cancer Institute, 1999. https://seer.cancer.gov/archive/pubncations/childhood/childhood-monograph.pdf (accessed 13 April 2022) [ Links ]
4. Birch JM, Blair V. The epidemiology of infant cancers. Br J Cancer 1992,66(Suppl 18).S2-S4. https://www.ncbijilm.nih.gov/pmc/articles/PMC2149659/pdf/brjcancersuppl00076-0006.pdf (accessed 4 February 2019). [ Links ]
5. Das U, AppajiL, KumariBSA, etal. A single center experience in 266 patients of infantile malignancies Pediatr Hematol Oncol 2014,31(6).489-497. https://doi.org/10.3109/08880018.2013.852644 [ Links ]
6. De Bouyn-Icher C, Minard-Colin V, Isapof A, Khuong Quang DA, Redon I, Hartmann O. Tumeurs solides malignes neonatales, à propôs de 71 cas. Arch Pediatr 2006,13(12).1486-1494. https://doi.org/10.1016/j.arcped.2006.08.014 [ Links ]
7. Alfaar AS, Hassan WM, Bakry MS, Qaddoumi I. Neonates with cancer and causes of death. Lessons from 615 cases in the SEER databases. Cancer Med 2017,6(7).1817-1826. https://doi.org/10.1002/cam4.1122 [ Links ]
8. Xue H, Horwitz JR, Smith MB, et al. Malignant solid tumors in neonates. A 40-year review. J Pediatr Surg 1995,30(4).543-545. https://doi.org/10.1016/0022-3468(95)90126-4 [ Links ]
9. Yang CP, Hung IT, Jaing TH, Shih LY, Chang WH. Cancer in infants. A review of 82 cases. Pediatr Hematol Oncol 2005,22(6).463-481. https://doi.org/10.1080/08880010591002233 [ Links ]
10. Rao S, Azmy A, Carachi R. Neonatal tumours. A single-centre experience. Pediatr Surg Int 2002,18(5-6)-.306-309. https://doi.org/10.1007/s00383-002-0720-8 [ Links ]
11. Bhatnagar SN. An audit of maügant solid tumors in infants and neonates. J Neonatal Surg 2012,1(1):5 http://www.elmedpub.comhttp//www.jneonatalsurg.com (accessed 7 February 2019). [ Links ]
12. Moore SW, Satgé D, Sasco AJ, Zimmermann A, Plaschkes J. The epidemiology of neonatal tumours. Report of an international working group. Pediatr Surg Int 2003,19(7).509-519. https://doi.org/10.1007/s00383-003-1048-8 [ Links ]
13. Chandrasekaran A. Neonatal solid tumors. Pediatr Neonatol 2018,59(1).65-70. https://doi.org/10.1016/j.pedneo.2016.12.007 [ Links ]
14. Kaatsch P. Epidemiology of childhood cancer. Cancer Treat Rev 2010,36(4).277-285. https://doi.org/10.1016/J.CTRV.2010.02.003 [ Links ]
15. Steliarova-Foucher E, Stiller C, Lacour B, Kaatsch P. International Classification of Childhood Cancer, third edition. Cancer 2005-,103(7).1457-1467. https://doi.org/10.1002/cncr.20910 [ Links ]
16. Hudson MM, Ness KK, Gurney JG, et al. Clinical ascertainment of health outcomes among adults treatedfor childhood cancer. JAMA 2013,309(22).2371-2381. https://doi.org/10.1001/jama.2013.629e [ Links ]
17. Brodeur GM, Pritchard J, Berthold F, et aL Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 1993,11(8).1466-1477. https:/doi.org/10.1200/JCO.1993.11.8.1466 [ Links ]
18. Clavien PA, Barkun J, de Oliveira ML, et al. The Clavien-Dindo classification of surgical complications. Five-year experience. Ann Surg 2009,250(2).187-196. https://doi.org/10.1097/SLA.0b013e3181bl3ca2 [ Links ]
19. Irtan S, Ehrlich PF, Pritchard-Jones K. Wilms tumor. 'State-of-the-arf update, 2016. Semin Pediatr Surg 2016,25(5).250-256. https://doi.org/10.1053/j.sempedsurg2016.09.003 [ Links ]
20. Honeyman JN.laQuagliaMP Malignant liver tumors. Semin Pediatr Surg 2012,21(3).245-254. https://doi.org/10.1053/j.sempedsurg2012.05.007 [ Links ]
21. Dasgupta R, Fuchs J, Rodeberg D. Rhabdomyosarcoma. Semin Pediatr Surg 2016,25(5).276-283. https://doi.org/10.1053/j.sempedsurg2016.09.011 [ Links ]
22. Dasgupta R, Rodeberg D. Non-rhabdomyosarcoma. Semin Pediatr Surg 2016,25(5).284-289. https://doi.org/10.1053/j.sempedsurg2016.09.012 [ Links ]
23. RescorlaFJ. Pediatric germ cell tumors. Semin Pediatr Surg 2012,21(1).51-60. https://doi.org/10.1053/j.sempedsurg.2011.10.005 [ Links ]
24. Jin M, Tian Z, Xie Y, et aL Diagnosis and treatment of infantile malignant solid tumors in Beijing China: A multicenter 10-year retrospective study Pediatr Investig 2020,4(3).178-185. https://doi.org/10.1002/ped4.12213 [ Links ]
25. Vasilatou-Kosmidis H. Cancer in neonates and infants. Med Pediatr Oncol 2003;41(1):7-9. https://doi.org/10.1002/mpo.10153 [ Links ]
26. Erdmann F, Kielkowski D, Schonfeld SJ, et al. Childhood cancer incidence patterns by race, sex and age for 2000 - 2006. A report from the South African National Cancer Registry Int J Cancer 2015-,136(11):2628-2639. https://doi.org/10.1002/ijc.29308 [ Links ]
27. Schickerling TM, Mackinnon D. Neonatal tumours. A single centre review S Afr J Oncol 2017,l.a24. https://doi.org/10.4102/sajo.vli0.24 [ Links ]
28. Hendricks M, Parkes J, Pillay K, et al. Outcomes of children with rhabdomyosarcoma treated with intensive chemotherapy, surgery, and radiotherapy through a period of protocol revision at a South African center, 1990 - 2010. Pediatr Blood Cancer 2017,64(3). https://doi.org/10.1002/pbc.26248 [ Links ]
29. Newman EA, Abdessalam S, Aldrink JH, et al. Update on neuroblastoma. J Pediatr Surg 2019,54(3) .383389. https://doi.org/10.1016/j.jpedsurg.2018.09.004 [ Links ]
30. Meyers R, Hiyama E, Czauderna P, Tiao GM. Liver tumors in pediatric patients. Surg Oncol Clin Ν Am 2021,30(2).253-274. https://doi.org/10.1016/j.soc2020.11.006 [ Links ]
31. Rhee DS, Rodeberg DA, Baertschiger RM, et al. Update on pediatric rhabdomyosarcoma. A report from the APSA Cancer Committee. J Pediatr Surg 2020,55(10).1987-1995. https://doi.org/10.1016/j.jpedsurg.2020.06.015 [ Links ]
32. Warmann SW, Seitz G, Fuchs J. Surgical complications in pediatric surgical oncology Pediatr Blood Cancer 2012,59(2).398-404. https://doi.org/10.1002/pbc24154 [ Links ]
33. Ullman AJ, Marsh N, Mihala G, Cooke M, Rickard CM. Complications of central venous access devices. A systematic review Pediatrics 2015,136(5).el331-el344. https://doi.org/10.1542/peds.2015-1507 [ Links ]
 Correspondence:
Correspondence:
N Jauquier
nicolas.]auquier@chuv.ch
Accepted 17 February 2022.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}