










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.112 no.1 Pretoria Jan. 2022
http://dx.doi.org/10.7196/SAMJ.2022.v112i1.16027
RESEARCH
Baseline characteristics of 32 patients with Gaucher disease who were treated with imiglucerase: South African data from the International Collaborative Gaucher Group (ICGG) Gaucher Registry
H SevitzI; F LaherII, III; S T VarugheseIV; M NelV; A McMasterVI; B F JacobsonVII, VIII
IMB BCh; Department of Obstetrics and Gynaecology, Faculty of Health Sciences, University of the Witwatersrand and Charlotte Maxeke Johannesburg Academic Hospital, Johannesburg, South Africa; Medi-Clinic Morningside, Johannesburg, South Africa
IIBDS; National Health Laboratory Service, Charlotte Maxeke Johannesburg Academic Hospital, Johannesburg, South Africa
IIIBDS; Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVMB BCh; Department of Paediatrics, Charlotte Maxeke Johannesburg Academic Hospital, Johannesburg, South Africa
VBPharm; Sanofi Genzyme, Johannesburg, South Africa
VIMB ChB; Sanofi Genzyme, Johannesburg, South Africa
VIIMB ChB, PhD; Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VIIIMB ChB, PhD Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND. Gaucher disease (GD) is a rare inherited autosomal recessive metabolic disorder with a prevalence in the general population of ~1 per 100 000. To optimise the recognition, diagnosis and management of patients with GD in South Africa (SA), it is important to have an understanding of local patterns of presentation of the disease.
OBJECTIVES. To describe the baseline pretreatment characteristics of the SA cohort of patients enrolled into the International Collaborative Gaucher Group (ICGG) Gaucher Registry whowere treated with imiglucerase (Cerezyme; Sanofi Genzyme).
METHODS. The ICGG Gaucher Registry is an observational, longitudinal, international database that tracks the clinical, demographic, genetic, biochemical and therapeutic characteristics of patients with GD globally, irrespective of disease severity, treatment status or treatment choice. The study population included all SA patients reported in the ICGG Gaucher Registry as of 1 May 2020.
RESULTS. The registry included 49 SA GD patients, of whom 32 received imiglucerase as first primary GD therapy. All the patients had GD type 1, 59.4% were female, and mean and median ages at diagnosis were 14.7 and 9.8 years, respectively. The most common genotype was N370S/N370S (37.5%). At treatment initiation, 30.0% of patients had been splenectomised. Among patients for whom data were available, anaemia was present in one-third of non-splenectomised patients and 12.5% of those with splenectomy, and moderate or severe thrombocytopenia was reported in two-thirds of non-splenectomised patients. Bone pain was present in 30.8% and 57.1% of non-splenectomised and splenectomised patients, respectively. No bone crises were reported, and data relating to other bone complications were available for only <3 patients.
CONCLUSIONS. Haematological findings and bone pain in this group are similar to those in the global ICGG Gaucher Registry cohort. Lack of baseline data for other bone complications limits interpretation in that regard. Clinicians who treat patients with GD are encouraged to submit accurate, complete and up-to-date information so that comprehensive data for the subset of SA GD patients can be maintained to improve recognition and diagnosis, and guide appropriate and effective use of treatment for SA patients.
Gaucher disease (GD) is a rare inherited autosomal recessive metabolic disorder, which if left untreated may be associated with significant morbidity and early mortality.[1] It is a lysosomal storage disease (specifically a sphingolipidosis) caused by mutations in the GBA1 gene located on 1q21 that codes for the enzyme acid beta-glucocerebrosidase. This lysosomal enzyme hydrolyses glucosylceramide into ceramide and glucose, but in GD its activity is markedly decreased, leading to accumulation of glucosylceramide in macrophages, which are hereafter referred to as Gaucher cells. In GD patients, residual enzyme activity ranges from 5% to 25% and, in rare cases, GD may be caused by mutations affecting the protein saposin C, an acid beta-glucocerebrosidase activator necessary for optimal enzyme activity.[1,2] Gaucher cells primarily infiltrate the bone marrow, spleen and liver, but can also infiltrate other organs, including the central nervous system. However, glucosylceramide and other sphingo- and phospholipids may also accumulate in the lysosomes of other phagocytic cells, outside of lysosomes in these and other cell types and in the plasma, and it is likely that a number of diverse processes, including a generalised systemic inflammatory response, contribute to the pathology and symptoms in patients with GD.[3-7]
Clinical phenotypes of GD represent a continuum of disease severity and progression, but are classically divided into three major subtypes (types 1, 2 and 3), which are differentiated depending on the presence or absence of primary central nervous system involvement, age of presentation and residual acid beta-glucocerebrosidase activity. Nevertheless, there is considerable phenotypic heterogeneity within each GD subtype.[8,9]
GD type 1 (GD1) is the most well-understood and well-characterised form of GD.[10] Patients with GD1 have some residual, but markedly diminished, acid beta-glucocerebrosidase activity. They do not have primary involvement of the central nervous system, but rather involvement of the hepatic, splenic, bone/bone marrow, haematological and pulmonary systems. The age of onset is variable. Although >50% develop symptoms before the age of 20 years, others become symptomatic only in adulthood or have minimal evidence of disease with few or no symptoms throughout their life.
Both liver and spleen may be massively enlarged. Hepatomegaly is a finding in 60 - 80% of patients, whereas splenomegaly is present in >90%, and asymptomatic splenomegaly is the most frequent presenting sign in GD1.[1] Hypersplenism and bone marrow infiltration are associated with pancytopenia (i.e. anaemia, leukopenia and thrombocytopenia), causing fatigue and increased risk of bruising and bleeding.[8,11] Clinical or radiographic evidence of bone disease occurs in 70 - 100% of individuals with GD1, ranging from asymptomatic osteopenia to focal lytic or sclerotic lesions and osteonecrosis. Acute and chronic bone pain, pathological fractures and subchondral joint collapse with secondary degenerative arthritis are a cause of considerable morbidity. Metaphyseal flaring, a deformity of the distal femur (Erlenmeyer flask deformity) as a result of undertubulation, may be visible on radiographs.[8,11] Bone infarction or bone crisis occurs in ~10% of subjects with GD and is especially common during childhood and adolescence.[8] Secondary neurological complications (e.g. spinal cord or nerve root compression) may occur consequent to bone disease or coagulopathy. In comparison with the general population, patients with GD may be at higher lifetime risk of several comorbidities, including Parkinson's disease (PD), insulin resistance and diabetes, and malignancy, especially multiple myeloma and haematological cancers.[12-15] Respiratory complications (e.g. pulmonary hypertension, interference with mechanical lung function) may occur secondary to splenomegaly and hepatic involvement, whereas infiltration of the lungs by Gaucher cells can lead to pulmonary fibrosis, although this latter finding is uncommon.[1,11] Data from the International Collaborative Gaucher Group (ICGG) Gaucher Registry have shown that compared with healthy individuals, life expectancy of those with GDI is shortened by ~9 years.[16]
The two neurological subtypes of GD are distinguished from each other based on the severity and rapidity of progression of neurological involvement. In GD type 2, early and severe neurological impairment is apparent by age 3 - 6 months and most patients die before 3 years of age. In comparison, juvenile or subacute neurological GD (type 3) is characterised by haematological and visceral signs and symptoms similar to those observed in type 1, combined with oculomotor neurological involvement. Disease onset is usually during childhood or adolescence, but approximately half present before 2 years of age. As in GDI, the presentation is heterogeneous. More severe neurological symptoms may include progressive myoclonus epilepsy, cerebellar ataxia or spasticity and dementia. In addition, there may be severe and progressive kyphosis, and the heart (with valve calcification) and cornea may be affected in some cases.[1]
Based on studies performed in North America and Western Europe, where GDI is the most common form, the prevalence of GD in the general population is estimated at 1 per 100 000. However, it is higher among Ashkenazi Jews, affecting ~1 in 855 individuals.[1,2,8]
Although the prevalence of GD type 3 in Europe, North America and Israel is estimated at ~5%, in some areas of the world, including the Philippines, Korea, Egypt, Japan and China, type 3 is a common GD phenotype, accounting for between 12% and 50% of individuals in local GD registries.[17-22]
Management of GD should be individualised. Goals of therapy are to prevent or improve haematological, visceral and bone symptoms; reduce (to normal if possible) liver and spleen volume; maintain normal growth, bone density and mobility; reduce fatigue; and maintain good quality of life and participation in normal daily activities and social and functional roles. Long-term goals include prevention and early detection of complications such as pulmonary disease, malignancy, parkinsonism/PD and diabetes.[23,24]
Although bone marrow transplantation has been used to treat GD in the past, it has been largely superseded by enzyme replacement therapy (ERT), which is the gold standard for management of GD, and substrate replacement therapy.[11] There are currently three recombinant acid beta-glucocerebrosidase ERTs: imiglucerase, velaglucerase alfa and taliglucerase alfa. When administered intravenously, imiglucerase has been shown to be safe and effective in reversing signs and symptoms resulting from haematological and hepatosplenic involvement, and in improving bone outcomes over 20 years of treatment.[25] Although they may improve quality of life in patients with GD1 and 3, ERTs do not alter the prognosis of neurological disease.[26]
Symptomatic management is now reserved for a small subset of patients who continue to have severe disease manifestations on ERT, or those who do not receive ERT. Although ERT has virtually eliminated the necessity for splenectomy, partial or total splenectomy may be considered in patients with massive splenomegaly, splenic infarction or severe thrombocytopenia. Other considerations include blood transfusions for severe anaemia and bleeding; analgesia for bone pain; joint replacement surgery for relief from chronic pain and restoration of function; and supplemental treatment with calcium and vitamin D for patients with low bone density.[11]
Because it is a rare disease, it is difficult to collect data on the natural history of GD, diagnosis, management, and the safety and efficacy of therapeutic regimens. The ICGG Gaucher Registry (NCT00358943) was established in 1991 to address this need. This registry is an observational, longitudinal, international database that tracks the clinical, demographic, genetic, biochemical and therapeutic characteristics of patients with GD globally, irrespective of disease severity, treatment status or treatment choice. It is the largest GD registry in the world, and since its inception has enrolled >6 000 patients with GD types 1, 2 and 3. The registry is supported by Sanofi Genzyme and is governed by a collaborative group of international physician experts in GD. The purpose of the registry is to collect observational longitudinal data from a real-world setting to describe phenotypic variability and the natural history of GD in untreated disease, age at presentation, indicators of disease and genotype. The data collected will help to guide recommendations for monitoring of patients, to evaluate short-, intermediate- and long-term effectiveness of treatment, and to inform hypothesis-driven research.[15,27]
To optimise the recognition, diagnosis and management of patients with GD in South Africa (SA), it is important to have an understanding of local patterns of presentation of the disease. In this article we describe the baseline pretreatment characteristics of an SA cohort of patients enrolled into the ICGG Gaucher Registry who were treated with imiglucerase.
Methods
The ICGG Gaucher Registry is a global observational database, which is governed by an international group of experts and supported by Sanofi Genzyme. It is open to all GD patients around the world, regardless of their treatment status or choice of treatment. Clinical assessments and care of patients enrolled in the registry are determined by their treating physicians. The only requirement for inclusion in the registry is a confirmed diagnosis of GD. Treating physicians voluntarily provide data on assessments currently used to monitor the clinical manifestations and disease progression of GD, and response to therapy.
At the request of the authors, data were provided by the Sanofi Genzyme Registry Team. The study population included all SA patients reported in the ICGG Gaucher Registry as of 1 May 2020. Demographic and clinical baseline characteristics reported to the registry were evaluated among the patients who received imiglucerase as first primary GD therapy. Because they are therapeutically equivalent, the term imiglucerase was used to refer to treatment with either imiglucerase (Cerezyme; Sanofi Genzyme) or alglucerase (Ceredase; Sanofi Genzyme).[28] Baseline was defined as the data point closest to imiglucerase initiation using a window of no more than -3 months/+2 weeks from treatment initiation for haemoglobin and platelet count, a window of no more than -6 months/+6 weeks from treatment initiation for liver and spleen volume, and a window of no more than -2 years/+6 weeks from treatment initiation for bone manifestations. Categorical variables were reported using frequencies and percentages, whereas continuous variables were described using frequencies, means, standard deviations, medians, 25th percentiles, 75th percentiles, and minimum and maximum values. To ensure patient privacy, data are not shown for subpopulations containing <5 patients, and where a genotype is reported for <5 patients, the frequency is reported as '<5'.
Results
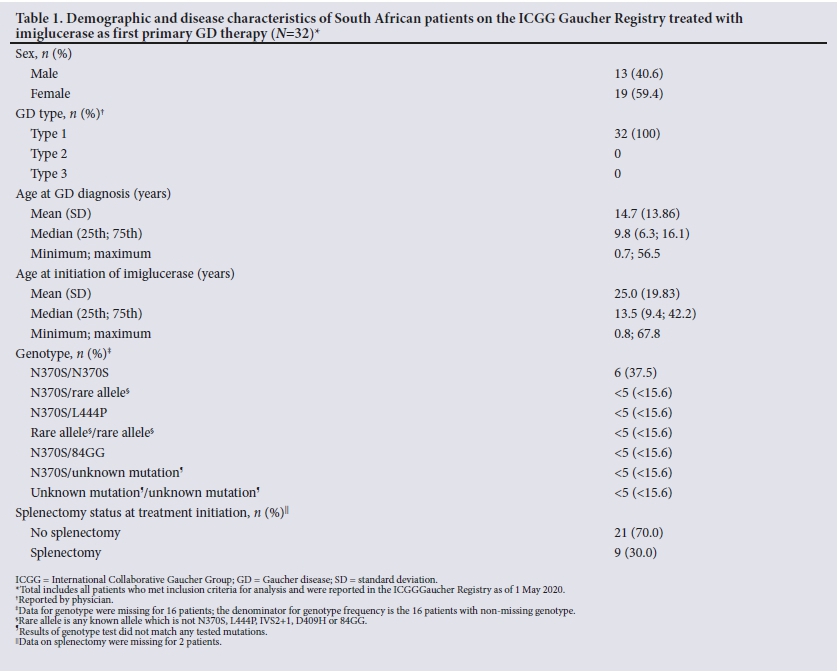
In total, as of 1 May 2020, the registry included 49 SA GD patients, of whom 4 were never treated, 32 received imiglucerase as first primary GD therapy, and 13 initiated treatment with an alternative agent. Demographic information for the imiglucerase treatment cohort is given in Table 1. All the patients had GDI, and 59.4% were female. The mean and median ages at diagnosis were 14.7 and 9.8 years, respectively. Genotype was documented for 16 patients, and the most common genotype was N370S/N370S (n=6/16; 37.5%). Other genotype subgroups included between 1 and 4 patients. Thirty percent (n=9/30) of the patients were splenectomised at treatment initiation.
The data availability at baseline for the 32 South African GD patients varied across parameters: 23 had haematological assessment, 3 had spleen volume assessment, 4 had liver volume assessment, and 30 had bone manifestation assessment (Table 2). At baseline, reported mean haemoglobin concentrations were 11.2 g/dL and 12.4 g/dL for patients with (n=15) and without (n=8) a spleen, respectively. While the majority of patients in both groups were not anaemic, the proportion of patients who had anaemia was greater among non-splenectomised patients than splenectomised patients (33.3% (n=5/15) and 12.5% (n=1/8), respectively). The mean platelet count was 112.7 χ 109/L and 288.9 χ 109/L among non-splenectomised and splenectomised patients, respectively. Moderate or severe thrombocytopenia (platelet count <120 χ 109/L) was reported in 66.6% (n=10/15) of non-splenectomised patients, whereas mild or no thrombocytopenia (platelet count >120 χ 109/L) was reported in all patients in the splenectomised group (n=8/8). Among the 3 and 4 patients with baseline data available for splenomegaly and hepatomegaly, respectively, moderate or severe splenomegaly (spleen size >5 times normal) was reported in 2 patients, and moderate or severe hepatomegaly (liver size >1.25 times normal) was reported in 3 patients.
Presence or absence of bone manifestations at baseline was reported for 21 patients in total. In non-splenectomised patients, bone pain was present in 30.8% (n=4/13) and absent in 69.2% (n=9/13). In the splenectomy group, bone pain was present in 57.1% (n=4/7) and absent in 42.9% (n=3/7). There were no patients with bone crises among patients with data available (n=0/20). Among 3 patients with marrow infiltration data available at baseline, marrow infiltration was present in 2 (n=1 splenectomised and 1 non-splenectomised). Data relating to avascular necrosis, bone infarction, fractures and Erlenmeyer flask deformity were available for only 1 patient each, all of whom were in the splenectomy group.
There were no data available at baseline for lytic lesions. Only 2 patients had data available relating to orthopaedic surgery. One had had 1 hip surgery, ~11 years before treatment initiation. The other patient had had 7 hip surgeries (joint replacement or revision) involving both hips, 3 before treatment initiation and 4 after. The first hip surgery was performed ~7 years before starting treatment for GD.
Discussion
The present study is the first description of the SA cohort in the ICGG Gaucher Registry. All the patients had GD1. The median age at diagnosis was 9.8 years, and the majority (75%) of the patients were diagnosed before 17 years of age. These figures are consistent with data from the overall international GD1 cohort, of whom the majority had onset (onset/diagnosis/recognition) in the first and second decades of life; 68% were diagnosed before age 10 years and only 28% after age 20 years.[10] The most frequent genotype in our cohort was homozygous N370S/N370S. Nevertheless, this genotype represents only approximately one-third of patients for whom genotyping was performed. Other genotypes reported were present in a limited number of patients (<5 per genotype) and were not further detailed, in order to preserve patient confidentiality. At the global level in the ICGG Gaucher Registry, a little over one-third of patients with the N370S allele are homozygotes, whereas the majority are heterozygous with the L444P, 84GG or another allele.[10] Previous studies have suggested that N370S/L444P is likely to be the most frequent genotype in GD1 populations of European descent, and generally this genotype leads to more severe disease compared with homozygous N370S.[24]
More than two-thirds of the treated patients in our cohort were not splenectomised at treatment initiation. The proportions of patients reporting anaemia (33% v. 12%) and moderate or severe thrombocytopenia (67% v. 0%) were greater among non-splenectomised v. splenectomised patients. The proportion of patients reporting bone pain was smaller among non-splenectomised patients compared with splenectomised patients (31% v. 57%). While numbers are small and should be interpreted with caution, this is not an unusual finding in GD. Splenectomy will ameliorate the cytopenia caused by hypersplenism, but it may be associated with worsening disease, including accelerated bone involvement, severe hepatic fibrosis and liver failure, lung infiltration and increased risk of infection.[3] The haematological findings in this group are very similar to those in the global ICGG Gaucher Registry cohort, of whom ~20 - 30% at baseline were anaemic and 50 - 70% were thrombocytopenic.[10]
The majority (n=20/32) of SA patients had bone pain and bone crisis assessment available at baseline. Among these, only 8 patients had bone pain present and no patients had bone crisis. Other bone manifestation assessments (i.e. marrow infiltration, avascular necrosis, infarction, Erlenmeyer flask deformity, fracture) at baseline were available for only a few patients (<3), which limits interpretation of these data. This finding contrasts with the global ICGG Gaucher Registry cohort of GD1 patients, among whom more than half reported bone pain, and radiographic evidence of avascular necrosis in any bone or evidence of infarction was found in ~10 -20%. Erlenmeyer flask deformities occurred in ~40% of all GD1 patients and in 63% of those aged <10 years at onset.[10] Fractures were recorded in 15%.[2] It is unclear whether this difference reflects a genuinely low incidence of these complications in the SA cohort, or under-reporting.
Goals of treatment for GD1 are to eliminate or reduce anaemia-related symptoms, bleeding and visceral complications, and to maintain mobility, general wellbeing and normal growth, and participation in school, work and social activities.[23] Management requires an individualised multidisciplinary approach, and ERT is the pharmacotherapeutic treatment of choice for symptomatic patients.[23,24,29-31] Imiglucerase is the most widely used ERT worldwide. It has revolutionised the treatment of GD and markedly improved the prognosis of patients with non-neuropathic forms of the disease, while virtually eliminating the need for therapeutic splenectomy.[25,26] We hope that this report of the SA cohort in the ICGG Gaucher Registry will contribute to the understanding of local patterns of GD and thereby help to improve recognition and diagnosis of patients with GD, and guide appropriate and effective use of treatment for SA patients.
Study limitations
This analysis has some limitations. Inherent to the analysis of a rare disease, there are a limited number of patients at a country level. Nevertheless, the ICGG Gaucher Registry provides the most extensive information available about GD patients in SA treated in a real-world setting. We have presented baseline data for 32 patients from SA sites in the Gaucher Registry who have been treated with imiglucerase, and the report excludes 4 untreated patients and 13 who were treated with non-imiglucerase therapy as primary treatment. Although we cannot comment on these excluded patients, we have included baseline data for approximately two-thirds of the SA cohort, which is a substantial proportion of the SA GD patients in the registry and is likely to be representative of the group as a whole.
Owing to the observational nature of the study, the registry is reliant on voluntary and accurate recording of data by physicians. Data availability varies across clinical practice. Although there is a recommended schedule of assessment, each physician is solely responsible for determining the appropriate clinical care for each patient.
Conclusions
In patients with GD, early diagnosis and appropriate treatment are essential for reducing complications, improving quality of life and avoiding inappropriate procedures.[26,32] In presenting the baseline data for this SA cohort of the ICGG Gaucher Registry, we aim to raise awareness of the condition. Given that the most common early manifestations of the disease are haematological, GD should be included in the differential diagnosis of patients with anaemia or thrombocytopenia where an alternative aetiology cannot be ascertained.
The observational registry operates under real-world clinical practice conditions and there are no imposed protocol visits or procedures. Furthermore, data entry in the registry is voluntary and depends on personnel resources at physician sites. As a result, data availability varies across geographical and clinical sites. We would encourage physicians who manage patients with GD to enrol their patients into the registry and to provide and maintain accurate, complete and up-to-date information so thatcomprehensive data for the subset of SA GD patients can be maintained. This will help to enhance understanding about GD and its management in SA, and thereby benefit thewhole GD community.
Declaration. None.
Acknowledgements. The authors thank Dr David Webb for writing assistance in preparation of the manuscript.
Author contributions. The authors contributed equally to the conception and design of the work; the acquisition, analysis and interpretation of data for the work; and drafting the work and revising it critically for important intellectual content. All authors approved the draft to be published and agreed to be accountable for all aspects of the work.
Funding. Sanofi paid costs related to writing the report.
Conflicts of interest. Data were provided by Sanofi Genzyme. HS received consultancy fees and travel grants from Genzyme/Sanofi, Shire/Takeda and Pfizer and is an investigator for the International Gaucher Registry. MN and AM are employees of Sanofi Genzyme. FL, BJ and STV report no conflicts of interest that are relevant to this publication.
References
1. Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 2017;18(2):441.https://doi.org/10.3390/ijms18020441 [ Links ]
2. Guggenbuhl P, Grosbois B, Chalés G. Gaucher disease. Joint Bone Spine 2008;75(2):116-124. https://doi.org/10.1016/j.jbspin.2007.06.006 [ Links ]
3. Deegan PB, Cox TM. Imiglucerase in the treatment of Gaucher disease: A history and perspective. Drug Design Dev Ther 2012;6:81-106. https://doi.org/10.2147/DDDT.S14395 [ Links ]
4. Dekker N, van Dussen L, Hollack CEM, et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011;118(16):e118-e127. https://doi.org/10.1182/blood-2011-05-352971 [ Links ]
5. Groener JEM, Poorthuis BJHM, Kuiper S, Hollak CEM, Aerts JMFG. Plasma glucosylceramide and ceramide in type 1 Gaucher disease patients: Correlations with disease severity and response to therapeutic intervention. Biochim Biophys Acta 2008;1781(1-2):72-78. https://doi.org/10.1016/j.bbalip.2007.11.004 [ Links ]
6. Fuller M. Gaucher's disease in the lipidomics era. Clin Lipidol 2012;7(4):431-441. https://doi.org/10.2217/clp.12.39 [ Links ]
7. Pandey MK, Burrow TA, Rani R, et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017;543:108-112. https://doi.org/10.1038/nature21368 [ Links ]
8. Burrow TA, Barnes S, Grabowski GA. Prevalence and management of Gaucher disease. Ped Health Med Ther 2011;2:59-73. https://doi.org/10.2147/PHMT.S12499 [ Links ]
9. Chen M, Wang J. Gaucher disease: Review of the literature. Arch Pathol Lab Med 2008;132:851-853. https://doi.org/10.5858/2008-132-851-GDROTL [ Links ]
10. Grabowski GA, Zimran A, Ida H. Gaucher disease types 1 and 3: Phenotypic characterisation of large populations from the ICGG Gaucher Registry. Am J Hematol 2015;90(S1):S12-S18. https://doi.org/10.1002/ajh.24063 [ Links ]
11. Pastores GM, Hughes DA. Gaucher disease. 2000 Jul 27 [Updated 2018 Jun 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle, Wash.: University of Washington, 1993 - 2020. https://www.ncbi.nlm.nih.gov/books/NBK1269 (accessed 8 September 2020). [ Links ]
12. Mistry PK, Taddei T, vom Dahl S, Rosenbloom BE. Gaucher disease and malignancy: A model for cancer pathogenesis in an inborn error of metabolism. Crit Rev Oncog 2013;18(3):235-246. https://doi.org/10.1615/critrevoncog.2013006145 [ Links ]
13. Utz J, Whitley CB, van Giersbergen PLM, Kolb SA. Comorbidities and pharmacotherapies in patients with Gaucher disease type 1: The potential for drug-drug interactions. Mol Genet Metab 2016;117(2):172-178. https://doi.org/10.1016/j.ymgme.2015.12.001 [ Links ]
14. Cox TM, Rosenbloom BE, Barker RA. Gaucher disease and comorbidities: B-cell malignancy and parkinsonism. Am J Hematol 2015;90(S1):S25-S28. https://doi.org/10.1002/ajh.24057 [ Links ]
15. Mistry PK, Belmatoug N, vom Dahl S, Giugliani R. Understanding the natural history of Gaucher disease. Am J Hematol 2015;90(S1):S6-S11. https://doi.org/10.1002/ajh.24055 [ Links ]
16. Weinreb NJ, Deegan P, Kacena KA, et al. Life expectancy in Gaucher disease type 1. Am J Hematol 2008;83(12):896-900. https://doi.org/10.1002/ajh.21305 [ Links ]
17. Schwartz IVD, Göker-Alpan Ö, Kishnani PS, et al. Characteristics of 26 patients with type 3 Gaucher disease: A descriptive analysis from the Gaucher Outcome Survey. Mol Genet Metab Rep 2018;14:73-79. https://doi.org/10.1016/j.ymgmr.2017.10.011 [ Links ]
18. Chiong MAD, Racoma MJC, Abacan MAR. Genetic and clinical characteristics of Filipino patients with Gaucher disease. Mol Genet Metab Rep 2018;15:110-115. https://doi.org/10.1016/j.ymgmr.2018.03.010 [ Links ]
19. Jeong SY, Park SJ, Kim HJ. Clinical and genetic characteristics of Korean patients with Gaucher disease. Blood Cells Mol Dis 2011;46(1):11-14. https://doi.org/10.1016/j.bcmd.2010.07.010 [ Links ]
20. El-Morsy Z, Khashaba MT, Soliman Oel-S, Yahia S, El-Hady DA. Glucocerebrosidase acid beta gene mutations in Egyptian children with Gaucher disease and relation to disease phenotypes. World J Pediatr 2011;7(4):326-330. https://doi.org/10.1007/s12519-011-0309-1 [ Links ]
21. Tajima A, Yokoi T, Ariga M, et al. Clinical and genetic study of Japanesepatients with type 3 Gaucher disease. Mol Genet Metab 2009;97(4):272-277. https://doi.org/10.1016/j.ymgme.2009.05.001 [ Links ]
22. Choy FT, Zhang W, Shi HP, et al. Gaucher disease among Chinese patients: Review on genotype/ phenotype correlation from 29 patients and identification of novel and rare alleles. Blood Cells Mol Dis 2007;38(3):287-293. https://doi.org/10.1016/j.bcmd.2006.11.003 [ Links ]
23. Biegstraaten M, Cox TM, Belmatoug N, et al Management goals for type 1 Gaucher disease: An expert consensus document from the European working group on Gaucher disease. Blood Cells Mol Dis 2018;68:203-208. https://doi.org/10.1016/j.bcmd.2016.10.008 [ Links ]
24. Mistry PK, Cappellini MD, Lukina E, et al A reappraisal of Gaucher disease - diagnosis and disease management algorithms. Am J Hematol 2011;86(1):110-115. https://doi.org/10.1002/ajh.21888 [ Links ]
25. Weinreb NJ, Camelo JS jr, Charrow J, McClain MR, Mistry P, Belmatoug N; for the International Collaborative Gaucher Group (ICGG) Gaucher Registry (NCT00358943) investigators. Gaucher disease type 1 patients from the ICGG Gaucher Registry sustain initial clinical improvements during twenty years of imiglucerase treatment. Mol Genet Metab 2021;132(2):100-111. [ Links ]
26. Gary SE, Ryan E, Steward AM, Sidransky E. Recent advances in the diagnosis and management of Gaucher disease. Expt Rev Endocrinol Metab 2018;13(2):107-118. https://doi.org/10.1080/17446651.2018.1445524. [ Links ]
27. Weinreb NJ, Kaplan P. The history and accomplishments of the ICGG Gaucher registry. Am J Hematol2015;90(S1):S2-S5. https://doi.org/10.1002/ajh.24054 [ Links ]
28. Grabowski GA, Barton NW, Pastores G, et al. Enzyme therapy in type 1 Gaucher disease: Comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med 1995;122(1):33-39. https://doi.org/10.7326/0003-4819-122-1-199501010-00005 [ Links ]
29. Bhengu L, Davidson A, du Toit P, et al. South African guidelines for the management of Gaucher disease, 2011. S Afr Med J 2012;102(8):697-702. https://doi.org/10.7196/SAMJ.5439 [ Links ]
30. Kaplan P, Baris H, de Meirleir L, et al Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr 2013;172(4):447-458. https://doi.org/10.1007/s00431-012-1771-z [ Links ]
31. Torralba-Cabeza M-A, Olivera-Gonzalez S, Sierra-Monzon J-L. The importance of a multidisciplinary approach in the management of a patient with type I Gaucher disease. Diseases 2018;6(3):69. https://doi.org/10.3390/diseases6030069 [ Links ]
32. Mistry PK, Weinthal JA, Weinreb NJ. Disease state awareness in Gaucher disease: A Q&A expert roundtable discussion. Clin Adv Hematol Oncol 2012;10(6 Suppl 8):1-16. [ Links ]
 Correspondence:
Correspondence:
M Nel
monique.nel@sanofi.com
Accepted 2 September 2021


{kind=link}
{kind=link}