










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.112 no.1 Pretoria Jan. 2022
http://dx.doi.org/10.7196/SAMJ.2022.v112i1.16011
IN PRACTICE
CASE REPORT
Gaucher disease: A cause of massive splenomegaly in a 15-year-old black African male
T RamdinI; E SchapkaitzII; S VarugheseIII; H SevitzIV; F LaherV; B F JacobsonVI
IMB BCh, MMed (Paed) Neonatol ; Department of Paediatrics and Child Health, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIMB BCh, MMed (Haem); Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIMB BCh; Department of Paediatrics and Child Health, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVMB BCh; Department of Obstetrics and Gynaecology, Charlotte Maxeke Johannesburg Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VBDS; Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VIMB ChB, PhD Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Patients with Gaucher disease (GD), a rare autosomal recessive lysosomal storage disease, commonly present to paediatricians with massive splenomegaly. While the diagnosis and management of patients with this chronic multisystem disorder has evolved significantly in recent years, the initial diagnosis represents a challenge. We describe the case of a 15-year-old black African male who presented with abdominal distension, delayed growth and fatigue. Initial laboratory studies revealed severe anaemia (haemoglobin concentration 8 g/dL) and moderate thrombocytopenia (platelet count 80 χ 109/L). A computed tomography scan of the abdomen showed an enlarged liver of 173 mm and massive splenomegaly of 27 mm. The diagnosis of GD was confirmed by reduced beta-glucocerebrosidase activity and heterozygous mutations in the GBA1 gene. The patient was managed at a dedicated paediatric haematology unit with enzyme replacement therapy and regular clinical, biochemical and radiological monitoring.
The paediatric patient with massive splenomegaly often poses a diagnostic challenge. While the list of conditions associated with splenomegaly is extensive, massive enlargement of the spleen is seen in few conditions.[1,2] In resource-limited countries, the most common cause is infection such as chronic malaria, visceral leishmaniasis and schistosomiasis. Lysosomal storage disorders represent a rare cause of massive splenomegaly. Gaucher disease (GD), the most common storage disorder, is caused by inherited mutations of the lysosomal enzyme glucocerebrosidase gene (GBA1), resulting in accumulation of the substrate glucocerebroside in the cells of the macrophage-monocyte system. Owing to its rarity, diagnostic delays can lead to life-threatening complications[3] We describe the investigation and management of a 15-year-old child with type 1 GD who presented with massive splenomegaly.
Case report
A 15-year-old boy of black African ethnicity presented with abdominal distention, loss of appetite, loss of weight and fatigue for a duration of 2 years. He had a significant travel history in the past 6 months to Mozambique. He had no significant past medical history or family history. On examination he was pale. On abdominal examination there was marked distension, a 21 cm non-tender splenomegaly and a 10 cm non-tender hepatomegaly (Fig. 1).

All other systems were within normal limits, including findings on neurological examination. The patient's weight and height plotted on the -2 z-score on the World Health Organization growth charts. He was admitted to an academic paediatric unit and was evaluated by haematology, infectious disease and genetics subspecialties.
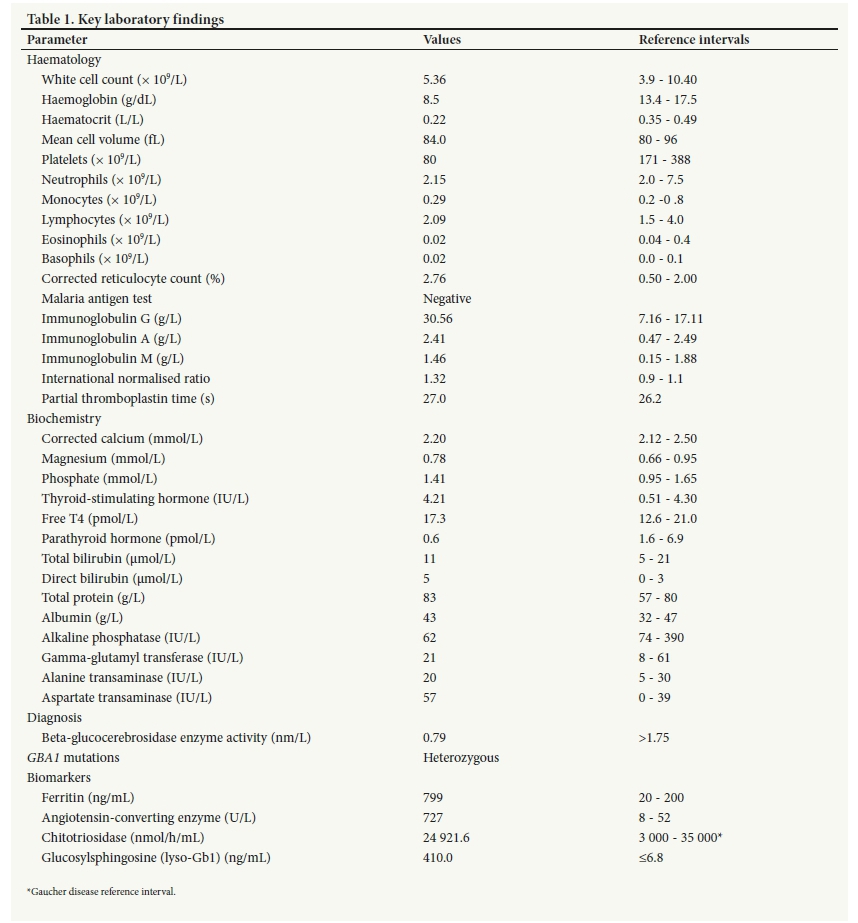
Initial laboratory studies revealed severe anaemia and moderate thrombocytopenia (Table 1). Radiological investigations included a chest radiograph, an ultrasound scan and a computed tomography (CT) scan of the abdomen. The ultrasound scan showed massive hepatosplenomegaly. There was reversed flow in the portal vein and splenic veins. CT showed an enlarged liver (173 mm) and spleen (27 mm) and a dilated portal vein (19 mm). Subsequently, liver biopsy and bone marrow biopsy were performed. Histological assessment of the liver revealed prominent macrophage infiltration of the sinusoidal and portal tracts. The bone marrow biopsy also showed increased macrophage activity. All lineages were present. Initially the presence of Gaucher cells was missed on the bone marrow biopsy specimen. On subsequent review, very occasional Gaucher cells were reported (Fig. 2.). A multiplex lysosomal storage disease screening assay was performed, and the sample was referred to an international genetics laboratory for genetic analysis. The diagnosis of GD was confirmed by reduced beta-glucocerebrosidase activity and heterozygous mutations in the GBA1 gene (new nomenclature c.222_224delp.(Thr75del) and c.259C>Tp.(Arg87Trp)).

The patient was transferred to a dedicated paediatric haematology unit. Enzyme replacement therapy with imiglucerase (Cerezyme; Sanofi Genzyme, USA) was commenced at the recommended dose of 40 U/kg every second week. The family was referred for genetic counselling. The patient's response to therapy was monitored regularly with clinical evaluation, laboratory tests including a full blood count, biomarkers of disease activity including chitotriosidase and glucosylsphingosine (lyso-Gbl), and radiological evaluation of the liver and spleen.
This study was approved by the Human Research Ethics Committee of the University of the Witwatersrand (ref. no. M200994).
Discussion
GD is a rare but important differential diagnosis for massive splenomegaly. The incidence of GD is ~1:50 000 - 1:100 000 in the general population. GD occurs in all ethnic groups in South Africa (SA), with a higher associated incidence in Ashkenazi Jews.[3] It is divided into three clinical types, of which type 1 (non-neuronopathic) accounts for the majority. All three types are caused by mutations n the GBA1 gene. More than 860 different mutations have been identified, which explains the various clinical presentations of GD.[4]
The majority of paediatric patients present with symptoms and signs in the first and second decades of life (Table 2).[5] Delayed diagnosis is characteristic for GD because the diagnosis is often not considered, leading to unnecessary investigations. The major systems involved in type 1 are the hepatic, splenic, skeletal, pulmonary and haematological systems. Absence of early-onset neurological involvement is necessary for the diagnosis of type 1. The clinical presentation and the severity of the disease are heterogeneous. Patients may be asymptomatic or, as in the case of the proband, present in childhood with significant clinical complications.!61 Fatigue has been described in 50% of patients, which may affect school performance. In children, delayed growth is also common. Splenomegaly, which is often massive, is observed in >90% of patients, but splenic rupture rarely occurs. Hepatomegaly is also noted in 60 - 80% of patients. At diagnosis, >80% of patients have signs of bone involvement affecting the bone marrow and/or structural compartments. This is one of the most disabling long-term complication of GD, often associated with acute painful bone crises, impaired mobility and reduced functionality. Mild mucocutaneous bleeding is also a common finding at presentation. Anaemia, which is usually moderate, is observed in 20 - 50% of patients and thrombocytopenia in 60 -90%. Leukopenia, however, is unusual. Rarely, patients present with interstitial lung disease caused by infiltration of the lungs by Gaucher cells.

In patients with the above clinical features of GD, the gold standard for diagnosis is demonstration of reduced beta-glucocerebrosidase activity in conjunction with whole GBA1 sequencing.[7, 8] Mutations that cause a truncated protein or an alteration in protein stability are associated with a more severe disease phenotype. Testing for specific biomarkers, such as chitotriosidase or glucosylsphingosine (lyso-Gb1), is helpful for diagnosis, disease monitoring in untreated patients, and monitoring a response to enzyme replacement therapy in treated patients. A bone marrow or liver biopsy are the incorrect diagnostic tests. Bone marrow findings of Gaucher cells are nonspecific. Cells resembling Gaucher cells, referred to as pseudoGaucher cells, can also be seen in various other haematological disorders. In SA, pseudo-Gaucher cells have also been reported in disseminated Mycobacterium tuberculosis and atypical mycobacterial infections. In our case, the diagnosis was initially missed because only occasional Gaucher cells were present. Unexplained hepatomegaly may, however, warrant a liver biopsy. Similarly, a bone marrow investigation may be indicated in atypical presentations as part of the differential diagnosis of childhood immune thrombocytopenic purpura or haematological malignancies.
Diagnostic imaging at presentation and on follow-up depends on the clinical presentation. An abdominal ultrasound scan is recommended for measurement of the liver and spleen size. A skeletal survey is indicated for an acute bone crisis or fracture.
Baseline dual-energy X-ray absorptiometry (DEXA) at the lumbar spine and femoral neck is valuable for assessment of osteopenia/ osteoporosis. A chest radiograph is the initial investigation for suspected pulmonary infiltration. Consultation with relevant specialists experienced in GD is fundamental for the management of these patients.
Initiation of disease-specific therapy in clinically significant GD is guided by presenting symptoms or deterioration over time. [9,10] The goals of treatment are to stabilise and improve clinical signs and symptoms and to prevent irreversible complications.[11] An additional important goal in children is to optimise growth.[5,6] Disease-specific therapy should be considered for the following indications: thrombocytopenia or anaemia, progressive hepatosplenomegaly, bone involvement, growth retardation, delayed puberty, pulmonary involvement, a history of splenectomy and impairment of functioning. Pre-symptomatic therapy can also be discussed in children with a family history of clinically significant complications. Genetic counselling plays an important role in helping patients and their families to understand the clinical condition, its management and the role of inheritance.
In SA, the treatment of choice for type 1 GD is recombinant imiglucerase (Cerezyme; Sanofi Genzyme Corporation, USA).[9,, 12] Imiglucerase was first introduced in 1994 and is a safe and effective treatment for the key disease characteristics of type 1 GD. However, this treatment is very costly, and in resource-limited settings children often struggle to attend one of the specialist facilities twice a month. The minimal effective dose should be initiated and thereafter dose adjustments are recommended in order to achieve and maintain therapeutic goals (Table 3). In patients with severe or high-risk criteria, the recommended dose is 30 - 60 U/kg administered by intravenous infusion over 1 - 2 hours every second week. Severe or high-risk criteria include symptomatic paediatric patients with severe genotypes, growth failure and/or delayed puberty, bone involvement, prior splenectomy, significant splenomegaly, hepatic complications, transfusion dependent/coagulopathy/platelets <20 χ 109/L, and pulmonary involvement. The recommended dose in patients who do not fulfil high-risk criteria is 10 - 30 U/kg every second week. Alternative forms of enzyme replacement therapy have recently become available in SA. Velaglucerase alfa, a gene-activated human glucocerebrosidase, is an alternative for poor responders (mainly due to allergies or neutralising antibodies). Oral substrate replacement treatments, which inhibit glucocerebroside synthase, are not registered in SA.[14] In asymptomatic patients with mild mutations, disease-specific therapy is often not warranted. However, regular monitoring is indicated for disease progression. In treated patients, regular monitoring is also imperative to ensure an adequate response to therapy.
Conclusions
Diagnostic delays occur frequently in GD and may result in severe complications such as growth failure, bleeding and bone pathology in children. Education of paediatricians is essential to improve early detection of patients with GD and facilitate the optimal management of this treatable disorder.
Teaching points
• Recognise the clinical features for the diagnosis of GD.
• Learn about the appropriate investigations for GD.
• Review the optimal management of GD.
Declaration. None.
Acknowledgements. None.
Author contributions. TR and ES: manuscript author; SV, HS, FL and BFJ: critical review of the manuscript; ES: histological examination. All authors read and approved the final manuscript.
Funding. None.
Conflicts of interest. None.
References
1. Pozo AL, Godfrey EM, Bowles KM. Splenomegaly: Investigation, diagnosis and management. Blood Rev 200933(3):105-111. https://doi.org/10.1016/j.blre.2008.10.001 [ Links ]
2. Kamble MB, Meena KB, Meena VK. Splenomegaly in children. Int J Contemp Pediatr 2019;6(4):1552-1556. https://doi.org/10.18203/2349-3291.ijcp20192753 [ Links ]
3. Elstein D, Abrahamov A, Hadas-Halpern I, Zimran A. Gaucher's disease. Lancet 2001;358(9278):324- 327. https://doi.org/10.1016/S0140-6736(01)05490-3 [ Links ]
4. Grabowski GA, Zimran A, Ida H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am J Hematol 2015;90(S1):S12-S18. https://doi.org/10.1002/ajh.24063 [ Links ]
5. Dinur T, Zimran A, Becker-Cohen M, et al. Long term follow-up of 103 untreated adult patients with type 1 Gaucher disease. J Clin Med 2019;8(10):1662. https://doi.org/10.3390/jcm8101662 [ Links ]
6. Weinreb NJ, Zimran A. The Gaucher Registry. Arch Intern Med 2000;160(18):2835-2843. https://doi.org/10.1001/archinte.160.18.2835 [ Links ]
7. Gary SE, Ryan E, Steward AM, Sidransky E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev Endocrinol Metab 2018;13(2):107-118. https://doi.org/10.1080/17446651.2018.1445524 [ Links ]
8. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 2008;372(9645):1263-1271. https://doi.org/10.1016/s0140-6736(08)61522-6 [ Links ]
9. Bhengu L, Davidson A, du Toit P, et al. South African guidelines for the management of Gaucher disease, 2011. S Afr Med J 2012;102(8):697-702. https://doi.org/10.7196/SAMJ.5439 [ Links ]
10. Zimran A. How I treat Gaucher disease. Blood 2011;118(6):1463-1471. https://doi.org/10.1182/blood-2011-04-308890 [ Links ]
11. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 2004;41(4 Suppl 5):4-14. https://doi.org/10.1053/j.seminhematol.2004.07.009 [ Links ]
12. Charrow J, Andersson HC, Kaplan P, et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: Consensus recommendations. J Pediatr 2004;144(1):112-120. https://doi.org/10.1016/j.jpeds.2003.10.067 [ Links ]
13. Biegstraaten M, Cox TM, Belmatoug N, et al. Management goals for type 1 Gaucher disease: An expert consensus document from the European working group on Gaucher disease. Blood Cells Mol Dis 2018;68:203-218. https://doi.org/10.1016/j.bcmd.2016.10.008 [ Links ]
14. Shemesh E, Deroma L, Bembi B, et al. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst Rev 2015, Issue 3. Art. No.: CD010324. https://doi.org/10.1002/14651858.CD010324.pub2 [ Links ]
 Correspondence:
Correspondence:
T Ramdin
tanusha.ramdin@wits.ac.za
Accepted 11 August 2021


{kind=link}
{kind=link}