










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.111 n.11 Pretoria Nov. 2021
http://dx.doi.org/10.7196/samj.2021.v111i11.15990
REVIEW
When cells become medicines: A South African perspective
IM ViljoenI; M S PepperII
IBPharm, BCom Hons; Institute for Cellular and Molecular Medicine, Department of Immunology, and SAMRC Extramural Unit for Stem Cell Research and Therapy., Faculty of Health Sciences, University of Pretoria, South Africa
IIMB ChB, PhD, MD, PD (Geneva); Institute for Cellular and Molecular Medicine, Department of Immunology, and SAMRC Extramural Unit for Stem Cell Research and Therapy., Faculty of Health Sciences, University of Pretoria, South Africa
ABSTRACT
The discovery of human leucocyte antigen (HLA), serological matching and HLA-typing techniques, combined with the development of immunosuppressive medicines and improvements in infection control, have opened the way to cell, tissue and vascularised organ transplantation. Since the early 1960s, more than a million haematopoietic progenitor cell (HPC) transplantations have been performed worldwide to restore haematopoiesis and support immune system recovery after bone marrow ablation. HPC transplantation uses minimally manipulated autologous or allogeneic cells to restore the homologous functions of bone marrow. Research in biological sciences supported by new technologies is increasingly translated into therapeutic products intended to augment, repair, replace or regenerate genes, cells, tissues, organs and metabolic processes in the body. These products are referred to as regenerative medicine therapies or advanced therapy medicinal products, and include gene therapies, cell-based therapies and engineered tissue products.
Cell-based therapies (CBTs) and engineered tissue products contain or consist of substantially manipulated living cells and tissues. CBTs are used to prevent, diagnose or treat disease through the pharmacological, immunological or metabolic action of living cells or tissues. Substantial manipulation or processing changes the biological characteristics, physiological functions or structural properties of the primary cells and tissues to fulfil new, nonhomologous functions in the recipient, and this level of manipulation differentiates manufactured CBTs from minimally manipulated cells or tissues of primary origin used for transplantation.
CBTs use living autologous or donated allogeneic cells or cells from cell banking systems as starting material. While research is ongoing into the use of human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs), the main research focus is on somatic cells, including HPCs, adoptive cell therapies (ACT) and mesenchymal stromal/stem cells (MSCs). In this article, we review some of the exciting products that have emerged from this research.
Stem cells and stem cell-like cells
In late 1998, several groups differentiated hESCs into the three germ layers, and further into various terminally differentiated cell types.[1,2]Ex vivo cultured stem cells could potentially offer benefits for identifying drug targets, preclinical research and toxicity studies for new drug leads. They also raised expectations of future therapies that could treat and cure inherited or acquired conditions such as cancer, heart disease, diabetes and Parkinson's disease. By 2001, hESC research became entangled in US biopolitics and bioethics, and federally funded research effectively ground to a halt. Teratoma formation presents a serious safety concern, and privately funded researchers are investigating knock-in genes that would allow teratoma elimination.[3,4]
In 2006, using a cocktail of four transcription factors (Oct3/4: Sox2, Klf4 and c-Myc), Takahashi and Yamanaka[5-7] reprogrammed mouse fibroblasts into pluripotent stem cells. The following year, they generated iPSCs from human fibroblasts. Autologous iPSC-derived cells and tissues hold promise for a wide range of transplantation and tissue engraftment applications, including haematopoietic stem cells, cells for wound repair and the repair or replacement of neurological and retinal cells, without the risk of immune rejection.[8,9] Early induction techniques relied on retroviral transfection to deliver transcription factors into somatic cells. This method carries the risk of viral genome integration. The use of non-integrating viral vectors such as Sendai virus, and adenovirus vectors, has been explored. Other methods, including plasmids, piggyBac vectors, minicircle vectors, episomal vectors, modified mRNA transcripts and protein transduction, have also been used.[10] The aim is to develop a safe, non-integrative, DNA-free method that is also efficient. Like hESCs, iPSCs carry a risk of iatrogenic tumorigenesis. The teratoma risk comes from the persistence of pluripotent cells in differentiated cell populations. Furthermore, c-Myc is an oncogene used for reprogramming, and its presence increases the risk of tumour formation.[11,12] A large amount of basic and non-clinical research still needs to be done before iPSC-derived products can reach the clinical trial stage.
Somatic cell-based products
Haematopoietic cells
Ex vivo haematopoietic cell expansion
HPC transplant outcomes can be improved by increasing the number of HPCs in the graft. Cord blood (CB) is an important source of HPCs as it provides an immediately available off-the-shelf product for patients in need of urgent transplantation. The HLA matching requirement of CB is also less stringent than for bone marrow and peripheral blood-derived haematopoietic cells, improving the chances of obtaining a match.[13] However, the limited number of total nucleated cells (TNCs) and CD34+ cells present in a CB unit (CBU) can lead to delayed engraftment, particularly in adults. The use of two CBUs is an option, but the high cost of CBUs is a constraint.
An alternative strategy is the ex vivo expansion of HPCs. A variety of expansion strategies are being explored. These include the use of Notch ligand,[14] mesenchymal stromal cell co-culture,[15] nicotinamide (NiCord),[16] copper chelation (StemEx),[17] StemRegenin-l[18] and UM171, a small molecule.[19]
A challenge in the manufacture of various expanded cell-based products is to ensure the correct combination of cell phenotypes in the final product. In the case of expanded CBUs, that means providing a mixture of both short-term haematopoietic progenitor cells to ensure rapid cell and immune recovery and long-term haematopoietic stem cells to ensure sustained haematopoiesis. One approach is to select the T-cells from the graft, expand only the CD34+ cells and then add T-cells back to the graft. Another challenge is to make these ex vivo-expanded cell-based products affordable. For this reason, companies are also developing larger-scale manufactured, off-the-shelf, allogeneic products that could be used for multiple patients.
Virus-specific T-cells (VSTs)
People are exposed to a variety of virus infections, including human herpesviruses (HHV-6 and -7), Epstein-Barr virus (EBV), BK virus and cytomegalovirus (CMV). CMV is common in low-and middle-income countries, and can be transmitted through bodily fluids, mother-to-child transmission (MTCT) during birth, through breast milk and through the placenta. Activated cytotoxic T-lymphocytes, also known as CD8+ T-cells, can keep CMV under control. However, they do not completely eradicate the virus. The virus becomes latent but persists in the body, and is reactivated when a person becomes immune-compromised or immune-suppressed.
In allogeneic HCT, the patient's T-cells are depleted after myeloablative therapy. In the absence of virus-specific CD8+ T-cells, reactivated viruses can present as different disease complications in transplantation patients. EBV, for example, can cause massive and often fatal lymphoproliferation. CMV can lead to CMV pneumonia, with a mortality of 50%, and lead to gastrointestinal tract ulcers, which can occur concurrently with graft v. host disease. Retinitis is another less common manifestation of CMV
From the late 1990s, several groups started to prepare virus-specific cytotoxic T-lymphocytes, and infused them prophylactically into EBV-positive patients, or used selected donor EBV-specific T-cells to treat EBV-positive lymphomas. However, the manufacturing process can take up to 3 months, and is not generally feasible for clinical application. When CBUs are used, most T-cells are naive, and this leaves patients susceptible to viral infections.[20,21] A method was developed that uses a 20% fraction of a CBU to manufacture triple-activated T-cells (CMV, EBV and adenovirus), and expands the cells to a mean of 1.47 x 108 cells within 28 days.[22]
Using naive CBUs, it might be possible to manufacture adequate cell doses of multi-VSTs as off-the-shelf products to prevent and treat viral infections after a single CBT
Adoptive cell therapies (ACT)
The body's immune system is its first line of defence against cancer However, cancers mutate and develop immune-evasive techniques. Adoptive cell therapies use a patient's immune cells, and multiply or modify them ex vivo to more effectively target cancers.
Tumour-infiltrating lymphocytes
An entire white blood cell repertoire including CD4+ and CD8+ T-cells, B-cells, natural killer (NK)-cells, dendritic cells, macrophages, neutrophils, basophils and eosinophils infiltrate tumours, recognise tumour cells and eliminate them. Some tumour cells are less immunogenic and can escape immunosurveillance. Tumour-infiltrating lymphocyte (TIL) therapies isolate a patient's T-cells that express tumour-targeting T-cell receptors (TCRs). The cell numbers are expanded ex vivo before readministering them. TIL therapy has successfully treated metastatic melanoma, and clinical trials are being conducted to treat lung, ovarian, bladder and breast cancer.
The traditional TIL manufacturing process is cumbersome. The patients tumour is resected and carefully dissected into small tumour fragments. The fragments are then typically cultivated in two 24-well plates in the presence of IL-2. The cancer cells eventually die, and the T-cells increase, with cells being split every 4 days for 5 weeks to a total of 60 - 100 24-well plates. TILs are then co-cultured with HLA-matched tumour cell lines, and interferon-y production is measured in each co-culture supernatant by enzyme-linked immunosorbent assay (ELISA). Positively selected cells are expanded in a rapid expansion protocol (REP) in T175 flasks and split into as many as 30 3-litre cell culture bags. After a further 2 weeks, cells are harvested, volume reduced, washed and prepared for infusion. The process can take up to 7 weeks, and is very labour intensive. At the time of writing, TIL therapy is still an investigative product, and is not approved by the Food and Drug Administration (FDA). Product development continues to optimise and shorten the manufacturing process and the manufacture of synthetic TILs with increased cytotoxicity, better tumour homing and less exhaustion through genetic engineering.[23,24]
Chimeric antigen receptor T-cells (CAR-T cells)
Tumours can present various cell-surface proteins or antigens that offer targets for cell-based treatments. Tumour-associated antigens (TAAs) such as CD 19 are proteins that display cell-linage specific expression patterns, and are normal cellular proteins overexpressed in tumours. Expression of TAA-targeting chimeric antigen receptors (CARs) can be induced in a variety of cell types, including macrophages, NK and T-cells, but the bulk of current CAR research and development is focused on the expression of CARs in T-cells.
CARs are transcribed from gene constructs delivered into host cells by vectors. The CAR consists of a single-chain variable fraction (scFv) from a monoclonal antibody that can target the tumour cell-surface antigen of interest, a linker, a transmembrane region, a costimulatory region(s) and a primary region. The construct can also contain other genes. First-generation CARs used a single CD3Ç primary stimulatory region and an Src-family tyrosine kinase such as ZAP-70. Second-generation CARs have an additional co-stimulation domain typically derived from either CD28 or 4-1BB. They recruit proteins that induce IL-2 production. IL-2 has several essential roles in the immune system. Third-generation CARs include a second co-stimulation domain, resulting in more cytotoxic activity on tumour cells. Fourth-generation CARs or 'T-cells redirected for universal cytokine killing' (TRUCKs) are second-generation CARs armed with an additional gene that can code for a cytokine to enhance anti-tumour activity. Fig. 1 shows the evolution of four generations or CARs.
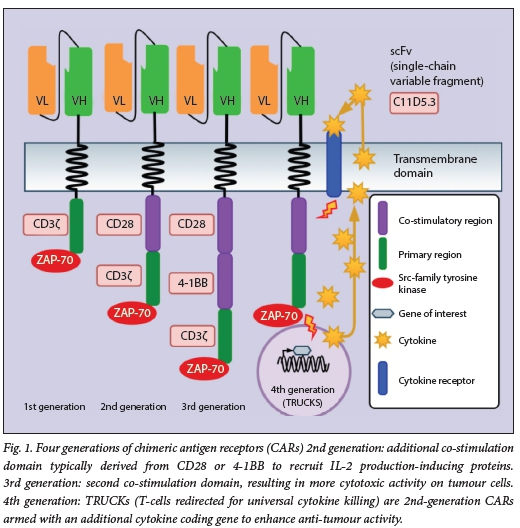
The efficacy of CARs is related to their ability to recognise tumour antigens and signal a response after receptor-ligand binding and cell activation, differentiation, proliferation and persistence. CAR-T cells must also have the ability to overcome immune system evasion by tumour cells. They can also elicit various adverse events, including cytokine release syndrome, neurotoxicity, on target/off tumour' recognition, anaphylaxis and cytopenia. Multiple approaches are being developed to enhance CAR selectivity, induce multiple CARs against different tumour cell-surface proteins including on/ off switches in the CAR-T cells, enhance tumour killing and enhance T-cell persistence, proliferation and survival.
CAR-T cell manufacture requires the collection of the patient's T-cells by apheresis, transporting the cells to a manufacturing facility where the cells are prepared, activated, transfected with a viral vector, expanded, harvested, packed and cryopreserved. The cell product is then shipped to a clinic where it is thawed and administered to the patient. The manufacturing process can take up to a month for a single dose. Fig. 2 shows (A) the steps in the collection, manufacturing and administration process, and (B) the transfection process using idecabtagene vicleucel (Abecma) as an example.

The first two CAR-T therapies, Tisagen-lecleucel (Kymriah, Novartis) and Yeskarta (axicabtagene ciloleucel, Kite Pharma), were licensed in late 2017 in the USA, and in 2018 in the European Union, for the treatment of refractory or relapsed B-cell acute lymphoblastic leukaemia in paediatric and young adult patients and relapsed or refractory diffuse large B-cell lymphoma in adults after two or more lines of systemic therapy. Both therapies target the CD19 cell-surface protein. In April 2021, the FDA approved lisocabtagene maraleucel (liso-cel) (Breyanzi) from Juno Therapeutics, a Bristol Myers Squibb company. Breyanzi also targets CD 19, and is likewise indicated for the treatment of diffuse large B-cell lymphoma. In 2020, Kite received both FDA and European Medicines Agency approval for Tecartus. Tecartus uses the same CAR construct as Yeskarta, but includes an additional manufacturing step to purify the harvested T-cells from circulating tumour cells.
During April 2021, the FDA approved Abecma (bluebird bio and Bristol Myers Squibb), a CAR-T-cell product indicated for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, which targets B-cell maturation antigen (BCMA). BCMA is also known as TNFRSF17 (tumour necrosis factor receptor superfamily member). Nearly all multiple myeloma tumour cells express BCMA, while normal tissue expression is restricted to plasma cells and a subset of mature B-cells.
Idecabtagene vicleucel is a second-generation CAR, manufactured from autologous peripheral blood mononuclear cells, stimulated with antibodies to CD 3 and CD28, and transduced with a lentiviral vector containing the anti-BCMA CAR, and expanded over a period of 10 days. The CAR is comprised of a murine extracellular scFv specific for recognising BCMA, attached to a human CD8 a hinge and transmembrane domain fused to the T-cell cytoplasmic signalling domains of CD 137 4-IBB and CD3-Ç chain, in tandem.[25]
Despite the risk of severe toxicity with CAR-t cell therapies, regulators approve the use of these therapies under a risk evaluation and mitigation strategy.
From a manufacturing perspective, viral vectors are manufactured in a biological process. The vector drug substance is then used to transfect the autologous cellular starting material to produce a new active substance. CAR-T cell products are, therefore, simultaneously gene-modified cell-based products and cell-based gene therapy. At the time of writing, ClinicalTrials.gov reported 510 active or recruiting clinical trials with CAR-T cell therapies in 389 conditions, mainly in liquid but also in solid tumours. The list also includes 33 allogeneic studies.[26]
Mesenchymal stromal/stem cells
Mesenchymal stromal/stem cells (MSCs) are spindle-shaped plastic-adherent cells that can be isolated from bone marrow, Wartons jelly adipose tissue and other tissue sources. MSCs have also been derived from iPSCs. Previously, these cells were incorrectly referred to only as mesenchymal stem cells. However, their 'sternness', or the ability of each cell to replicate and differentiate, could not be confirmed. It is now believed that a subpopulation of cells within the 'stromal' population has the ability to differentiate into a limited number of tissue-specific cell types. The term 'stem cells' is, however, still frequently used by researchers, laypersons and so-called 'stem cell clinics' that sell unproven treatments. Various cell surface antigens have been used to define MSCs phenotypically. In 2006, the International Society for Cell Therapy (ISCT) proposed minimal criteria to define multipotent MSCs as (i) being plastic adherent; (ii) expressing CD73, CD90 and CD 105; (iii) lacking the expression of CD34, CD45, CD lib, CD 14, CD 19, CD79a and HLA-DR haematopoietic and endothelial markers; and (iv) having the capacity to differentiate in vitro into adipocyte, chondrocyte and osteoblast lineages. MSCs are a heterogeneous cell population that includes fibroblasts and myofibroblasts, and may include a small proportion of stem or progenitor cells.[27,28] Due to the heterogeneity of MSCs derived from various sources, the ISCT updated their recommendation in 2019, requiring that the cell source should be used in conjunction with the MSC acronym. [29,30]
MSCs are heterogenous at many levels. In addition to the cell source, cells can be autologous or allogeneic. The cells can be used as isolated MSCs, MSCs plus carrier systems or MSC derivatives such as MSC-derived iPSCs and extracellular vesicles, to name some. In addition, there may be cell-processing centre-related heterogeneity in terms of sampling, production, equipment used, operator skill and potency assays. Finally, there may be significant recipient-related heterogeneity and heterogeneity related to the clinical application. The ability to definitively characterise MSCs remains challenging, and no single cell surface marker has been identified that can be used to identify these cells as 'MSCs'.
Initial research on MSCs focused on their differentiation into different phenotypes, but more recent research has been conducted on their immune-modulatory effects, their ability to migrate to sites of injury and their paracrine effects, mediated through their secretóme of bioactive molecules. The secretóme influences the microenvironment, providing cytoprotection, promoting tissue repair and stimulating angiogenesis.
MSCs represent attractive opportunities for their regenerative and immunomodulatory properties. A survey of cellular and tissue-engineered therapies in Europe in 2016 and 2017 reported 227 research teams treating 8 236 patients, with MSCs constituting 56% (4 416) of treatments, and HCs 21%. Of the MSC therapies, 24% of treatments formed part of clinical trials, and 6% were case studies. Seventy percent of respondents claimed that the treatments were 'routine therapies'. This is concerning, as there is currently no evidence base to support offering MSCs as routine therapies, and there are still no authorised MSC therapies in Europe. The sources of MSCs used were bone marrow (56%), adipose tissue (20%), cord blood (11%) and placenta (10%).[31,32]
Discussion
Cell-based regenerative medicine therapies (RMTs) represent a novel class of health products that hold the promise of addressing various unmet medical needs. They are different from primary cells and tissues used for transplantation, but also from conventional small-molecule and biopharmaceutical molecule-based medicines. These differences include the complexity and variability of cellular starting materials, the use of raw materials and reagents of biological origin, disruptive manufacturing technologies, the scale of manufacture, living cells and competent vectors as active components, limited patient numbers with complex disease profiles and the invasive nature of treatments. It is essential that legislative and regulatory frameworks should accommodate the unique nature of these therapies. For this purpose, various jurisdictions have modified existing legislation and added new regulations or have constructed lex specialis to provide for the regulation of this novel class of health product. Starting in 1993, the FDA published a notice in the Federal Register on how it intended to apply its current statutory authorities governing therapeutic products to human somatic cell therapy products and gene therapy products. The European Union followed in 2007, Australia in 2011 and Japan in 2014. Canada, Singapore, Taiwan, South Korea, Brazil, Russia, India and China have followed more recently, developing their own legal and regulatory frameworks to support national priorities.
The South African (SA) National Health Act No. 61 of 2003 (NHA)[33] and its regulations,[3436] which regulate the use of human biological materials, and the SA Medicines and Related Substances Act No. 101 of 1965[37] have not kept pace with scientific developments in the field of RMTs and manufactured cell-based products. This has created a regulatory vacuum that on the one hand negatively impacts on bona fide local research and development, as well as manufacture and access to advanced cell-based products, but on the other also allows so-called 'stem cell clinics' to sell unproven therapies, exploiting desperate and vulnerable patients.[38,39]
This legal and regulatory gap needs to be addressed urgently to protect SA patients from exploitation, and to guide and support SA researchers in developing RMTs for the benefit of SA patients.
Declaration. This article was written to partly fulfil the requirements of a PhD in Medical Immunology.
Acknowledgements. None.
Author contributions. IMV conceptualised, researched and wrote the original draft. MSP co-conceptualised the overarching thematic and reviewed and edited the manuscript throughout.
Funding. This work was funded by the South African Medical Research Council (Extramural Unit for Stem Cell Research and Therapy) and the University of Pretoria through the Institute for Cellular and Molecular Medicine.
Conflicts of interest. None.
References
1. Thomson JA. Embryonic stem celllines derived from human blastocysts. Science 1998;282(5391):1145-1147. https://doi.org/10.1126/science.282.5391.1145 [ Links ]
2. Shamblott MJ, Axelman J, Wang S, et al. Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc Nat Acad Sci 1998;95(23):13726-13731. https://doi.org/10.1073/pnas.95.23.13726 [ Links ]
3. Baker M. Why hES cells make teratomas. Nat Rep Stem Cells 2009:36. https://doi.org/10.1038/stemcells.2009.36 [ Links ]
4. He J, Rong Z, Fu X, Xu Y. A safety checkpoint to eliminate cancer risk of the immune evasive cells derived from human embryonic stem cells. Stem cells 2017;35(5):1154-1161. https://doi.org/10.1002/stem.2568 [ Links ]
5. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126(4):663-676. https://doi.org/10.1016/j.cell.2006.07.024 [ Links ]
6. Yamanaka S. Induction of pluripotent stem cells from mouse fibroblasts by four transcription factors Cell Proliferation 2007;41:51-56. https://doi.org/10.1111/j.l365-2184.2008.00493.x [ Links ]
7. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131(5):861-872. https://doi.org/10.1016/j.cell2007.11.019 [ Links ]
8. Takahashi K, Okita K, Yamanaka S, et al. Generation of retinal cells from mouse and human-induced pluripotent stem cells. Neuroscience Letters 2009;458(3):126-131. https://doi.org/10.1016/j.neulet2009.04.035 [ Links ]
9. Sarkar A, Saha S, Paul A, et al Understanding stem cells and its pivotal role in regenerative medicine Life Sci 2021;273:119270. https://doi.org/10.1016/j.lfs.2021.119270 [ Links ]
10. Gorecka J, Kostiuk V, Fereydooni A, et al. The potential and limitations of induced pluripotent stem cells to achieve wound healing. Stem Cell Res Ther 2019;10:87. https://doi.org/10.1186/sl3287-019-1185-1 [ Links ]
11. Wan W, Cao L, Kalionis B, Xia S, Tai X. Applications of induced pluripotent stem cells in studying the neurodegenerative diseases. Stem Cells Int 2015:382530. https://doi.org/10.1155/2015/382530 [ Links ]
12. Bedel A, Beliveau F, Lamrissi-Garcia I, et al. Preventing pluripotent cell teratoma in regenerative medicine applied to hematology disorders. Stem Cells Translational Medicine 2016;6(2):382-393. https://doi.org/10.5966/sctm.2016-0201 [ Links ]
13. Viijoen IM, Hendricks CL, Meliet J, Pepper MS. Perspectives on establishing a public cord blood inventory in South Africa. Cytotherapy 2021;23(6):548-557. https://doi.org/10.1016/j.jcyt.2021.02.116 [ Links ]
14. Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. Notch-medi ate d expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nature Med 2010;16(2):232-236. https://doi.org/10.1038/nm.2080 [ Links ]
15. Mehta RS, Saliba RM, Cao K, et al. Ex vivo mesenchymal precursor cell-expanded cord blood transplantation after reduced-intensity conditioning regimens improves time to neutrophil recovery. Biol Blood Marrow Transplant 2017;23(8):1359-1366. https://doi.org/10.1016/j.bbmt.2017.05.002 [ Links ]
16. Horwitz ME, Chao NJ, Rizzieri DA, et al. Umbilical cord blood expansion with nicotinamide provides long-term multilineage engraftment. J Clin Investig Am Soc Clin investig 2014;124(7):3121-3128. https://doi.org/10.1172/jci74556 [ Links ]
17. Stiff PJ, Montesinos P, Peled T, et al. Cohort-controlled comparison of umbilical cord blood transplantation using carle cortemcel-1, a single pro genitor-enriched cord blood, to double cord blood unit transplantation. Biol Blood Marrow Transp 2018;24(7):1463-1470. https://doi.org/10.1016/j.bbmt.2018.02.012 [ Links ]
18. Wagner JE, Brunstein CG, Boitano AE, et al. Phase lili trial of Stem Re genin-1 expanded umbilical cord blood hematopoietic stem cells supports testing as a stand-alone graft. Cell Stem Cell 2016;18(1):144-155. https://doi.org/10.1016/j.stem.2015.10.004 [ Links ]
19. Fares I, Chagraoui J, Lehnertz B, et al. EPCR expression marks UM171-expanded CD34+ cord blood stem cells. Blood Am Soc Hematol 2017;129(25):3344-3351. https://doi.org/10.1182/blood-2016-11-750729 [ Links ]
20. Lucas K, Small T, Heller G, Dupont B, O'Reilly R. The development of cellular immunity to Epstein-Barr virus after allogeneic bone marrow transplantation. Blood Am Soc Hematol 1996;87(6):2594-2603. https://doi.org/10.1182/blood.v87.6.2594.bloodjournal8762594 [ Links ]
21. Koehne G, Smith KM, Ferguson TL, et aL Quantitation, selection, and functional characterization of Epstein-Barr virus-specific and alioreactive T cells detected by intracellular interferon-y production and growth of cytotoxic precursors. Blood Am Soc Hematol 2002;99(5):1730-1740. https://doi.org/10.1182/blood.v99.5.1730 [ Links ]
22. Abraham AA, John TD, Keller MD, et ai. Safety and feasibility of vir us-specific T cells derived from umbilical cord blood in cord blood transplant recipients. Blood Adv Am Soc Hematol 2019;3( 14):2057-2068. https://doi.org/10.1182/bloodadvances.2019000201 [ Links ]
23. Hopewell EL, Cox C, Prion-Thomas S, Keliey LL. Tumor-infiltrating lymphocytes. Streamlining a complex manufacturing process. Cytotherapy 2019;21(3):307-314. https://doi.org/10.1016/j.jcyt.2018.11.004 [ Links ]
24. Jiménez-Reinoso A, Nehme-Alvarez D, Domínguez-Alonso C, Alvarez-Vallina L. Synthetic TILs*. Engineered tumor-infiltrating lymphocytes with improved therapeutic potential. Front Oncol 2021;10:593848. https://doi.org/10.3389/fonc.2020.593848 [ Links ]
25. Friedman KM, Garrett TE, Evans TW, et al. Effective targeting of multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther 2018;29(5):585-601. https://doi.org/10.1089/hum.2018.001 [ Links ]
26. ClinicalTrial.gov. Recruiting, active, not recruiting studies. CAR-T list results. ClinicalTrials.gov, 2021. https://clinicaitriais.gov/ct2/results?recrs=ad&cond=CAR-T (accessed 12 October 2021). [ Links ]
27. Horwitz EM, Le Blanc K, Dominici M, et al. Clarification of the nomenclature for MSC. International Society for Cellular Therapy position statement. Cytotherapy 2005;7:393-395. https://doi.org/10.1080/14653240500319234 [ Links ]
28. Wolmarans E, Mellet J, Durandt C, Joubert F, Pepper MS. Single-cell transcriptome analysis of human adipose-derived stromal cells identifies a contractile cell subpopulation. Stem Cells Int 2021;2021:1-12. https://doi.org/10.1155/2021/5595172 [ Links ]
29. Dominici M, le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement Cytotherapy 2006;8(4):315-317. http://doi.org/10.1080/14653240600855905 [ Links ]
30. Viswanathan S, Shi Y, Galipeau J, Nolta J, Phinney DG, Sensebe L. Mesenchymal stem versus stromal cells, international Society for Cellular Therapy Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019;21(10):1019-1024. https://doi.org/10.1016/j.jcyt.2019.08.002 [ Links ]
31. Gay MH, Baidomero H, Passweg J, Martin I. The survey on cellular and tissue-engine ere d therapies in Europe and neighboring Eurasian countries in 2016 and 2017. Cytotherapy 2020;22(5):S165. https://doi.org/10.1016/j.jcyt.2020.03.347 [ Links ]
32. Martin I. Presentation of Cellular and Tissue Engineered Therapies Survey. New Orleans. ISCT Annual Meeting, 2021. [ Links ]
33. South Africa. National Health Act No. 61 of 2003. As amended by Act No. 12 of 2013 and proclaimed in Proclamation 38, Government Gazette 37730,10 June 2014. [ Links ]
34. National Department of Health, South Africa. Regulations relating to the use of human biological materiaL R177, Government Gazette 35099,2 March 2012. [ Links ]
35. National Department of Health, South Africa. Regulations relating to blood and blood products. Government Notice R179. Government Gazette 35099,2 March 2012. [ Links ]
36. National Department of Health, South Africa. Regulations relating to stem cell banks. Government Notice R183. Government Gazette 35099,2 March 2012. [ Links ]
37. South Africa. Medicines and Related Substances Act No. 101 of 1965. As amended. Government Gazette 41009,28 July 2017. [ Links ]
38. Pepper MS. Partial relief from the regulatory vacuum involving human tissues through enactment of chapter 8 of the National Health Act and regulations thereto. S Afr Med J 2012;102(9):736-737. https://doi.org/10.7196/samj.5940 [ Links ]
39. Skeen M, Eksteen C, Pepper M. Stem cell tourism and spinal cord injury in South Africa. S Afr Med J 2019;109(8b):17-23. https://doi.org/10.7196/samj.2019.v109i8b.13824 [ Links ]
 Correspondence:
Correspondence:
IM Viljoen
igviljoen@gmail.com
Accepted 3 August 2021

