










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.111 n.9 Pretoria Sep. 2021
http://dx.doi.org/10.7196/SAMJ.2021.v111i9.15898
CME
Thalassaemia (part 2): Management
N A AlliI; M PatelII; J PooleIII; Y GogaIV; F FazelV; N NovitzkyVI; S ParasnathVII; F BassaVIII
IMB BCh, FCPath Haem (SA); Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
IIMB ChB, FCP (SA), MMed (Int Med), FRCP (London), PhD; Department of Medicine, Chris Hani Baragwanath Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIMB BCh, DCH (SA), FCP (SA) Paed; Department of Paediatrics and Child Health, Paediatric Haematology/Oncology Unit, Faculty of Health Sciences, University of the Witwatersrand and Charlotte Maxeke Hospital, Johannesburg, South Africa
IVMB BCh, DCH (SA), FCPaeds (SA), Cert Clin Haem Paed (SA), MSc Bioethics, PG Dip Paed Pall Medicine; Department of Paediatrics and Child Health, Nelson R Mandela School of Medicine, University of KwaZulu-Natal and Inkosi Albert Luthuli Central Hospital, Durban, South Africa
VMB BCh, FCP (SA), MMed (Int Med), Cert Clin Haem (SA); Blood Centre, Rondebosch Medical Centre, Cape Town, South Africa
VIDip Med, PhD; Division of Clinical Haematology, Department of Internal Medicine, Faculty of Medicine and Health Sciences, Tygerberg Academic Hospital and Stellenbosch University, Cape Town, South Africa
VIIMB ChB, FCPath (SA), Cert Clin Haem (SA); Department of Clinical Haematology, Inkosi Albert Luthuli Central Hospital and Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
VIIIMB ChB , FCPath Haem (SA), MMed (Haem); Division of Clinical Haematology, Department of Internal Medicine, Faculty of Medicine and Health Sciences, Tygerberg Academic Hospital and Stellenbosch University, Cape Town, South Africa
ABSTRACT
The management of thalassaemia with a severe phenotype includes blood transfusion, iron chelation, bone marrow transplantation, prenatal diagnosis and national programmes to co-ordinate these in countries with a high prevalence. If blood transfusion and iron chelation therapy are not administered regularly, as was the case historically and as is still the case in many poorer regions, progressive deterioration occurs, viz. impaired growth and development, hepatosplenomegaly, bony abnormalities, cardiac failure, increased susceptibility to infections and premature mortality. Remarkable progress has been made in the past few decades, which has led to much-improved survival rates. Transfusion therapy has evolved to a hyper-transfusion regimen designed to maintain a physiological haemoglobin level and achieve a posttransfusion haemoglobin of 14 g/dL, which, as a matter of course, necessitated intensification of iron chelation. The development of effective oral iron chelators has led to improved compliance. Exploration of novel therapeutic approaches continues, with several agents under study. The prospect of gene therapy is particularly exciting as it has potential to provide cure on a large scale. Currently, regular blood transfusion and iron chelation therapy remain the cornerstone of management of thalassaemia major.
Classification, pathophysiology and diagnosis of thalassaemia are discussed in part of this 2-part CME series. The current article (part 2) discusses management aspects relating to the three clinical phenotypes, viz.: (i) thalassaemia major (TM), also referred to as transfusion-dependent thalassaemia (TDT); (ii) thalassaemia intermedia (TI), which is generally transfusion independent; and (iii) thalassaemia minor, which is asymptomatic, with no significant clinical sequelae.
Thalassaemia major
The management of TDT major requires appropriate transfusion therapy, frequent monitoring to assess iron overload, and treatment with focused chelation therapy. With these in place, patients will survive longer, without the morbidity of complications due to inadequate chelation. Transfused blood contains iron (Fe), which the body is unable to excrete. Regularly transfused patients accumulate iron at a rate of 0.3 - 0.6 mg/kg of body weight per day.[2] Once iron is >12 - 24 g of total body iron, it accumulates in the heart, liver and endocrine system.
Blood transfusion
Once the diagnosis has been made, the infant needs to be monitored clinically with regular full blood counts (FBC).
The decision to commence regular transfusions[2] is based on the following criteria:
• A haemoglobin (Hb) level <7 g/dL on two occasions, >2 weeks apart (excluding all other contributory causes such as infections); or
• A steady state Hb <7 g/dL with any one of the following:
• failure to thrive
• bone expansion or deformity, especially facial bones
• inappropriate fatigue
• poor feeding
• developmental delay or regression
• cardiac failure
• increasing splenomegaly
• In most cases, it may be reasonable to administer the first transfusion, then wait and re-assess the indications and trends before the second and subsequent transfusions.
• Patients with TI should not receive regular transfusion therapy unless indicated. The diagnosis of TI and TM is not always absolute or predictable, and a period of watching and waiting may be necessary.
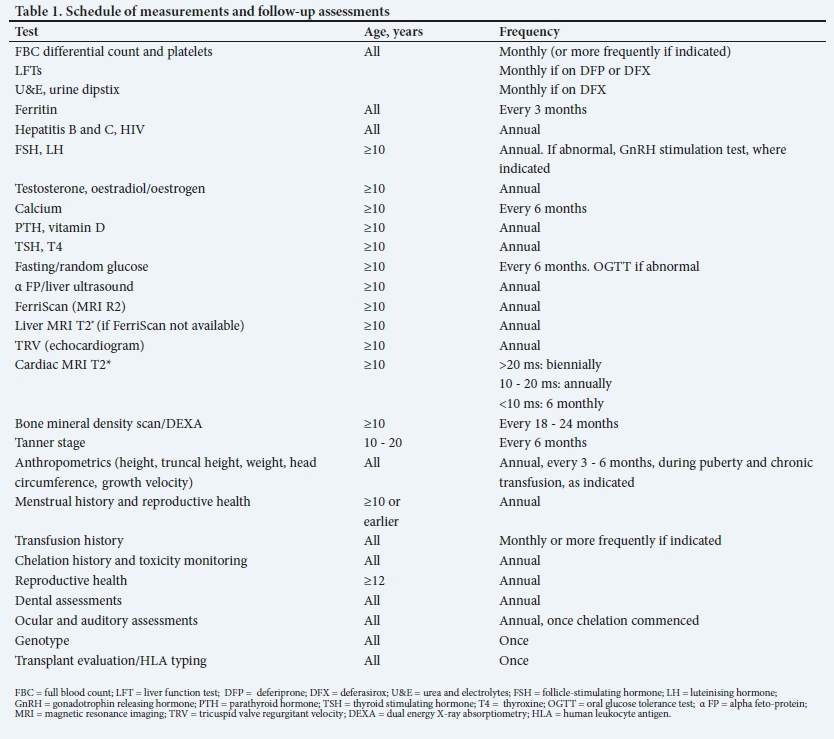
Before starting blood transfusions, baseline tests need to be performed, which includes an FBC, red cell phenotype, liver function tests (LFTs), serum iron studies, serum ferritin and baseline viral determination for hepatitis B (HBV), hepatitis C (HCV), HIV, cytomegalovirus (CMV) and parvovirus B19. Regular follow-up with blood tests at selected intervals is important for early identification of complications (Table 1).
Transfusion programme
Transfusion therapy aims to permit normal growth and activity levels, and prevent skeletal changes due to marrow hypertrophy. Transfusions should initially be given every 3 - 5 weeks.[3]
The pre-transfusion Hb determines the volume of blood to be transfused, and the aim is to maintain the pre-transfusion Hb between 9 g/dL and 10.5 g/dL.
In paediatric patients, transfusion volume (mL) = (target Hb 14 g/dL - actual Hb) χ weight (kg) χ 3.5. The post-transfusion Hb should not exceed 14 g/dL. Blood should be transfused at a rate of 5 mL/kg/hour.
In patients with severe anaemia (Hb <5 g/dL) or those with cardiac compromise, transfusions should be administered at 2 mL/kg/h to avoid fluid overload. Diuretics may need to be administered.
If cardiac insufficiency is present, pre-transfusion Hb levels need to be maintained at higher levels, i.e. between 11 g/dL and 12 g/dL, and small volume transfusions may need to be administered weekly or every 2 weeks.
Transfusion volume should be recorded at each visit, and the transfusion requirements reviewed 6 - 12-monthly. The usual transfusion requirement is between 200 mL/kg/year and 250 mL/kg/year.
Ideally, the type of blood product to be used is leukodepleted packed red cells, preferably <5 days old. Some blood banks can supply highly packed red cells by a process of spinning and aspirating excess plasma.
Blood transfusions should be given in a dedicated facility, and administered with minimal disruption to the patient, to maintain good quality of life. Good transfusion practice should be observed with written policies (standard operating procedure).
Iron overload and chelation therapy
Regularly transfused patients accumulate iron at a rate of 0.3 -0.6 mg/kg of body weight per day.[2] The human body does not have a physiological mechanism to excrete iron. When total body iron exceeds 12 - 24 g, it accumulates in the heart, liver and endocrine system.
Iron is extremely toxic to many tissues. Excess iron that exists as non-transferrin bound iron (NTBI) in the plasma and unbound iron in the intra-cellular compartment results in free radical damage to cells. It takes 3 - 10 years of chronic exposure to high iron levels before any measurable organ dysfunction occurs. Each organ has a different rate of iron deposition and reduction.[2-9]
The most effective strategy to prevent significant iron overload and tissue damage is early institution of chelation therapy, i.e. as soon as the patient is iron-overloaded.
Chelation therapy has two goals: (i) binding of toxic NTBI in the plasma; and (ii) removal of iron from the body.
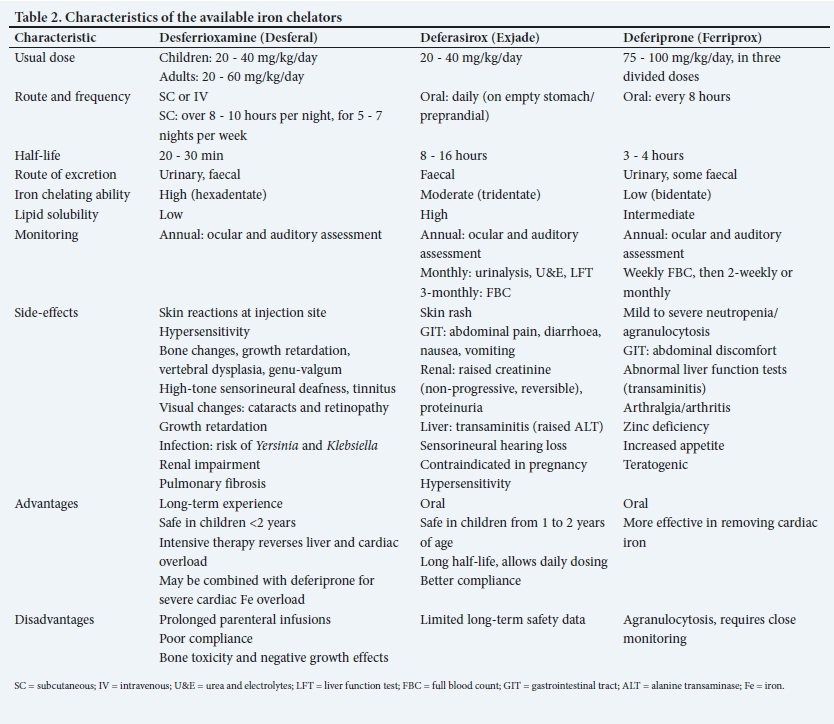
Since removal of iron from normal tissues can be detrimental, chelation should not be started without evidence of iron overload. In infants, chelation therapy should be delayed until after the first year of life due to the toxicity of the chelators. It is generally commenced after 10 - 12 transfusions, or when the ferritin is persistently >1 000 μg/L. First-line therapy is usually oral deferasirox (DFX), which can be commenced in an infant >1 year of age. Subcutaneous administration of desferrioxamine (DFO) is an alternative but more traumatic for both patients and parents/ caregivers, given the parenteral route of administration and longer duration of administration.
The aims of iron chelation therapy are:[2-9]
(i) to prevent iron overload and maintain a safe level of body iron by balancing iron intake from transfusion with iron excretion through chelation.
(ii) rescue therapy: once excess iron accumulates in organs, damage starts to occur. Removal of iron is slow and inefficient, therefore prevention is superior to rescue therapy.
(iii) emergency therapy: if heart failure develops, this requires urgent modification of chelation therapy with intensive treatment (see below).
There are currently three iron chelators available in South Africa (SA), viz. DFO mesylate (DFX) (Desferal), deferasirox (Exjade) and deferiprone (DFP)(Ferriprox). DFP availability is limited, and it currently requires a section 21 application via the SA Health Products Regulatory Authority. Characteristics of each iron chelator are detailed in Table 2.
All chelation regimens aim to attain and maintain an annual average serum ferritin of 500 μg/L - 1 000 μg/L, a liver iron between 3 mg/g and 7 mg/g dry weight (dw) and cardiac T2* >20 ms2.
The selection of drugs and doses will require adjustments depending on the iron burden and drug toxicities (Fig. 1).

Iron overload needs to be monitored regularly. Direct measurement of liver iron concentration via liver biopsy is invasive.
The following are methods of indirect measurement of tissue iron stores:
• serum ferritin
• magnetic resonance imaging (MRI) T2* for cardiac iron assessment
• MRI R2 (FerriScan) for liver iron assessment.
Serum ferritin is simple to perform, inexpensive and widely available. It is most useful in identifying trends. Levels should be monitored 3-monthly after the first 10 transfusions. Decisions to modify dosages should be based on serial measurements of ferritin as opposed to a single reading. Persistently high levels >2 500 ug/L are associated with increased risk of cardiac disease and death. Ferritin levels correlate well with liver iron levels in a linear fashion, but do not always correlate well with myocardial iron deposition, as patients with low ferritin levels may have severe cardiac iron loading and left ventricular (LV) impairment.
Liver iron: The R2 technique (FerriScan) can be performed on a standard MRI machine, but the analysis requires specific software. The advantage of the T2* technique is that liver and cardiac iron can be measured at the same time. The target should be 3 - 7 mg/g dw equivalent measured by the FerriScan technique. Liver iron >7 mg/g dw is associated with increased complications, including a higher risk of cardiac disease and early death.
Cardiac iron: The best available method for the measurement of cardiac iron is the cardiac MRI T2*. There is a relationship between low T2* and impaired LV function. LV impairment becomes increasingly likely when T2* falls below 20 ms, and is therefore used as a cut-off for the early detection of iron overload.
Cardiac MRI T2* and the FerriScan are recommended for routine monitoring in children from age >10 years. The intervals of the scans depend on whether the results are abnormal or normal (Fig. 1).
Chelation therapy needs to be modified when there is evidence of severe iron overload and/or organ dysfunction. This may be achieved with the following strategies:
(i) Combination therapy:'[2] combination treatment with DFO and DFP has been shown to be more effective than DFO monotherapy in: a) reducing serum ferritin; b) reducing liver iron; c) improving cardiac function; d) reversing cardiac failure; and e) reversing impaired glucose tolerance.
(ii) Optimising dosing or switching of preparations.
(iii) Intensive intravenous chelation with continuous high-dose DFO: this is used when aggressive chelation is required with severe iron overload and cardiac or liver dysfunction. This can be given twice a month for a minimum of 72 hours at a maximum dose of 6 g/kg/day, and is given in addition to the regular chelation therapy.
Chelation therapy also needs to be modified if body iron falls or reaches low levels, and doses need to be adjusted to prevent toxicity. Doses should be gradually decreased when the serum ferritin falls below 1 000 μg/L. Although the current recommendation is to stop the drug when the ferritin is <500 μg/L, one must be cautious, as this may result in rebound labile iron. This can be avoided by gradual dose reductions coupled with more intensive monitoring (monthly ferritin levels) to maintain the ferritin between 500 μg/L and 1 000 μg/L.
Management of complications
Iron overload is the major cause of morbidity and mortality in TM/TI, and occurs rapidly in patients on a chronic transfusion programme. Most deaths occur due to iron-related cardiomyopathy. Cardiac iron loading takes 8 - 10 years to develop, and the heart is initially devoid of iron for years. Once iron loading in the heart starts, the rate of deposition is rapid.
Iron overload also affects the liver, resulting in cirrhosis and liver failure. Iron deposition in the liver occurs in a linear fashion, and significant iron loading ensues after 6 months of regular transfusions. Pancreatic, thyroid, parathyroid, hypothalamic and pituitary damage may result in endocrinopathies.
Hepatic complications
Liver disease in thalassaemia is common, and frequently multi-factorial.[10] The liver is a primary site of iron overloading, and is one of the leading causes of mortality in TDT. Chronic liver iron deposition stimulates fibrogenesis and cirrhosis.[11]
Patients may present with acute or chronic hepatitis (viral or drug-induced), obstructive jaundice, cholangitis, portal hypertension, liver insufficiency or hepatocellular carcinoma. Chronic active HCV infection should be actively managed. The current recommended treatment for HCV is with a combination of pegylated interferon alpha and ribavirin. Ribavirin may result in haemolysis with increased transfusion requirements and iron chelation while on the drug.
Serum ferritin and liver iron deposition have a good correlation. Assessment of liver iron was previously done via a direct measure of dry liver iron and required a needle biopsy or intraoperative wedge biopsy. This is invasive, and as liver iron deposition is patchy, the results are poorly reproducible. MRI is a sensitive measure of tissue iron levels and is a non-invasive alternative to biopsy. Liver iron should be assessed by R2 or T2* MRI annually. Aggressive intensification of chelation should be instituted if liver iron concentration is >7 mg/g dw. Regular screening for liver dysfunction is recommended (Table 2), which includes monthly LFTs if patients are on DFX or DFP. If the alanine transaminase is increased, it should be repeated after 2 weeks, and if it remains elevated, or is intermittently elevated over a 3-month period, a complete assessment for hepatitis would be indicated[12] viz.,
• international normalised ratio/partial thromboplastin time
• hepatitis A IgM
• hepatitis B DNA quantification
• hepatitis C Ab and polymerase chain reaction
• CMV serology, viral load and liver biopsy if the hepatitis C is positive
• ultrasound (US), computed tomography (CT) and endoscopic retrograde cholangio-pancreatogram (ERCP), as indicated.
Hepatitis B vaccination should be given, and antibody titres checked annually to assess immune status. Booster vaccinations may be required (Table 3).
Liver complications should be managed in conjunction with a hepatologist.
Cardiovascular complications
Cardiac disease due to thalassaemia includes LV dysfunction, pulmonary hypertension and right ventricular (RV) dysfunction.[16] The mechanism of LV dysfunction includes a high-output cardiac state, iron overload, vascular disease, myocardial ischaemia and myocarditis.[6] Effective iron chelation reduces the risk of cardiac disease and improves survival. Cardiac iron overload develops from age 10 years in poorly chelated patients. Serum ferritin and liver iron concentration levels do not correlate well with cardiac iron overload.[16] Cardiac MRI T2* is the best available method for the detection of cardiac iron overload. There is a relationship between low T2* and impaired LV function. LV impairment becomes increasingly likely when T2* falls below 20 ms. T2* is used to monitor response to chelation, which should be adjusted as necessary.
Early identification of cardiac abnormalities and prompt treatment, including aggressive chelation, can reverse cardiac damage. Small-volume blood transfusions (to avoid fluid overload) given more frequently, such as fortnightly, may be needed. Higher target pre-transfusion Hb level (10 - 12 g/dL) is recommended if there is cardiac dysfunction.[4]
Splenectomy, transfusion intensity (frequency and pre-transfusion Hb), history of thrombosis and severity of iron overload are the strongest predictors of pulmonary hypertension. These patients can be effectively managed by initiating regular transfusions to maintain a pre-transfusion Hb of 9.5 g/dL. Thromboprophylaxis should be considered in patients with severe pulmonary hypertension.
Echocardiography and electrocardiograms (ECGs) should be performed annually from age 10 years. Patients with cardiac failure and arrhythmias require urgent cardiac assessment and management. If the tricuspid regurgitant velocity (TRV) is 2.5 - 3.2 m/s and asymptomatic, transfusions and iron chelation should be optimised, and hydroxyurea and anticoagulant therapy should be considered. TRV >3.2 m/s requires referral to the cardiologist for further management.
Endocrine complications
Iron is commonly deposited in endocrine organs, and damage that occurs is usually irreversible. This may result in diabetes mellitus (DM), growth failure, hypothyroidism, hypoparathyroidism, delayed puberty, hypogonadism, gonadal failure and sterility, as well as osteopenia and osteoporosis. The pituitary gland is particularly sensitive to even modest iron overload, with short stature in poorly chelated patients presenting around age 10 years. Regular monitoring of endocrine function is important (Table 1). Growth retardation and skeletal changes may also ensue from DFO toxicity. Patients with a fall-off on growth curves, decreased height velocity or delayed bone age should be referred to an endocrinologist for assessment and management. Hormone replacement therapy is advised for adolescents with hypogonadism.[31
Infections
Patients with iron overload are susceptible to infection with bacteria such as Yersinia enterocolitica and Vibrio vulnificus. Yersinia is a Gram-negative bacillus that causes severe infection and disseminated tissue sepsis. V. vulnificus is a Gram-negative bacillus that can cause severe endotoxaemia, and is associated with high mortality.[171
Other bacteria such as Escherichia coli and Klebsiella pneumoniae also occur with increased frequency in the presence of iron overload.
Despite significant improvements in blood safety, there remains a risk of transfusion-transmitted infections (TTI). The four major TTIs are caused by HIV, HBV, HCV and syphilis.
Routine paediatric vaccinations should be kept up to date, including the annual influenza vaccine.
Bone complications
Bone disease in thalassaemia results from bone marrow expansion, iron overload, iron chelation, endocrine dysfunction and vitamin D, C and K and zinc deficiencies.
The clinical presentation includes growth impairment, ricketslike bony lesions, back pain, spinal deformities, signs of nerve compression, severe osteoporosis and fragility fractures. Long bones, especially the humeri, may be short, with loss of the concave profile, and signs of growth arrest.[18]
Timeous and adequate transfusion therapy will prevent irreversible bony abnormalities that occur due to bone marrow expansion, viz. dental abnormalities, malocclusion, recurrent sinusitis and increased susceptibility to fractures.
Paediatric patients on DFO may develop rickets-like bony lesions and sclerotic lesions, severe epiphyseal dysplasia resulting in genu valgum or genu varum, short stature, truncal shortening and vertebral dysplasia.!18' These skeletal abnormalities are associated with early initiation of DFO or high doses of DFO in the presence of reduced iron stores. In the latter scenario, skeletal changes are preventable by using lower doses of DFO (20 - 30 mg/kg/d). Bony changes have not been associated with the oral iron chelators, such as DFP and DFX. However, DFP may result in arthropathy and is associated with bone mineralisation defects, hypercalciuria and nephrocalcinosis.
There is a high prevalence of vitamin D deficiency, and this may contribute to osteopenia. Vitamin D supplementation should be commenced when levels are suboptimal. Established osteoporosis in adults should be treated with bisphosphonates and managed by an endocrinologist. Zinc supplementation should be instituted in those with low zinc levels.
Renal disease
Renal disease in thalassaemia is a less well-recognised complication, caused by the disease itself, as well as iron chelators. Manifestations include tubular dysfunction, glomerular dysfunction and resultant proteinuria, haematuria, nephrolithiasis and renal failure.[19] Up to 60% of TDT patients develop tubular dysfunction, which is related to iron overload, chronic anaemia and DFO toxicity. A renal biopsy is invasive and risky; therefore, MRI is essential for non-invasive assessment of iron overload. There is also a significant correlation between serum ferritin and renal iron deposition. Splenectomy is an independent risk factor for tubular abnormalities.
DFX is very lipophilic and thus enters the tubular cells to form a highly charged complex with iron, causing a proximal tubulopathy and Fanconi syndrome.[19]
Thromboembolic disease
TM may be considered a hypercoaguable state, resulting from abnormalities in platelets and pathological red cells, endothelial dysfunction, coagulation system abnormalities and the presence of microparticles. Splenectomy in patients >35 years of age, anaemia (Hb <9 g/dL) and iron overload all contribute to the hypercoagulable state. Patients should be treated according to standard local thrombosis management guidelines.
Splenectomy in TM
(see appendix 1: http://samj.org.za/public/sup/15898.pdf).
Thalassaemia in pregnancy
(see appendix 2: http://samj.org.za/public/sup/15898.pdf).
Supportive care[3] Psychological support
Thalassaemia patients and their parents require strong social circles and regular psychological support through various stages of the disease, including diagnosis, initiation of transfusion and iron chelation, and transition to self-care.
Psychosocial factors strongly influence adherence to treatment and can also predispose to illnesses such as depression. Ongoing support and encouragement by a multidisciplinary team including a psychologist and social worker are essential to ensure continued adherence to treatment and to address any psychosocial issues.
Dental care
Patients with thalassaemia are vulnerable to many dental complications, including malocclusion, an increased risk of dental caries and periodontal disease. Regular dental follow-up is necessary.
Lifestyle and quality-of-life issues
With increasing life expectancy of thalassaemia patients, healthcare providers should try their best to ensure that the disease and its treatment cause the least amount of disruption as possible to patients' daily lives. Clinic appointments and transfusion regimens should be arranged in such a way that they have minimal impact on school or work activities.
Physical activity and sport should be encouraged, bearing in mind limitations due to anaemia and cardiopulmonary complications of the disease.
Attention to adequate nutrition includes monitoring for zinc and vitamin D deficiency and encouraging a diet rich in calcium and vitamin E. L-carnitine supplements may have beneficial antioxidant and cardioprotective effects.
Education, prevention and counselling
Patients need to be provided with age-appropriate information and educational tools regarding their disease, its complications and treatment.
Genetic counselling
This should be available for couples who wish to have a child, and options for prenatal diagnosis must also be discussed.
Transition to adult care
Transition to self-care and adult clinics coincides with adolescence. This is a period when patients will challenge boundaries, and adherence is compromised. The transition must be gradual, and only when the patient is emotionally ready. Holding joint paediatric and adult outpatient clinics allows for a smoother transition to adult care, and is less intimidating to the patient.
Novel therapeutic approaches
New therapeutic strategies aimed at improving the quality of life and improving outcomes are evolving as a result of improved understanding of the pathophysiology of β-thalassaemia. These include the following:
Addressing ineffective erythropoiesis
Growth differentiation factor 11 (GDF-11): this is overexpressed in immature erythroblasts in β-thalassaemia and induces expansion of erythroid progenitors with consequent ineffective erythropoiesis.[20] Novel strategies to bind/trap GDF11 include sotatercept and luspatercept.
In the BELIEVE trial studying the use of luspatercept, 21% of adults showed a 33% reduction in transfusion requirements, compared with 4.5% on placebo.[20,21]
Janus kinase 2 (JAK2) inhibitors: preclinical studies of JAK2 inhibition in mouse models showed improvement in ineffective erythropoiesis and a decrease in splenomegaly, suggesting that the use of ruxolitinib (a JAK1/JAK2 inhibitor) could benefit ß-thalassaemia patients.[22,23] The TRUTH Study in TDT showed a decrease in spleen size but no clinically significant improvements in pre-transfusion Hb, hence did not proceed into a phase III study.[24]
Improving iron overload
Minihepcidins: hepcidin is known to prevent iron utilisation and absorption. Preclinical studies propose that minihepcidins could restrict iron absorption and may be useful in addressing iron overload and improve ineffective erythropoiesis, anaemia and splenomegaly.[22,24]
Transmembrane protease serine 6 (TMPRSS6) inhibitors: TMPRSS6, a transmembrane serine protease produced by hepatocytes, negatively regulates hepcidin expression (by cleaving haemojuvelin) and is thus an important modulator of hepcidin expression.
Manipulation of ferroportin inhibitors and HIF2a: ferroportin plays a crucial role in iron homeostasis by exporting iron from enterocytes to the circulation. Intestinal HIF2a (hypoxia-inducible factor-2a) regulates ferroportin expression in response to changes in systemic iron requirements.[22]
Haemoglobin F (fetal haemoglobin) inducers
Haemoglobin F inducers include: (i) DNA methylation inhibitors, such as 5-azacytidine and decitabine; (ii) hydroxyurea; and (iii) short-chain fatty acids.
Bone marrow transplantation
(see appendix 3: http://samj.org.za/public/sup/15898.pdf).
Gene therapy
(see appendix 4: http://samj.org.za/public/sup/15898.pdf).
Thalassaemia intermedia
TI or non-transfusion dependent thalassaemia (NTDT) refers to the clinical phenotype of thalassaemia where patients do not require regular lifelong blood transfusions for survival.[25] This group includes a-thalassaemia intermedia (HbH disease), ß-thalassaemia intermedia and mild to moderate forms of HbE/ß-thalassaemia.[26]
Patients with TI present with anaemia with varying degrees of severity. Ineffective erythropoiesis, chronic anaemia and iron overload can lead to more severe clinical complications in TI than in a routinely transfused, well-chelated patient with ß-thalassaemia major[27] (see Table 3, part 1, CME[1] for clinical complications of thalassaemia).
Patients with TI should be on routine folic acid supplementation. The specific management of patients with TI includes conventional modalities, novel agents and stem cell transplantation.
Transfusion therapy
Patients with TI are generally transfusion independent. They may require blood transfusions during certain clinical situations, such as pregnancy, surgery or infection.[28] Selected patients may also be instituted on a more regular transfusion regimen for the primary or secondary prevention of specific clinical complications, such as pulmonary hypertension, thromboembolic events, leg ulcers and extramedullary haematopoiesis.[29]
Iron chelation
Iron overload in TI results from increased intestinal iron absorption as a result of chronic hypoxia. Serum ferritin levels underestimate the degree of iron overload in NTDT,[30,31] but are nonetheless still a useful test to guide chelation therapy in resource-constrained settings where MRI may not be available. Ferritin levels should be monitored 3-monthly, starting at the age of 10 years, and chelation therapy instituted or interrupted at levels of >800 μg/L and <300 μg/L, respectively). Based on available evidence, DFX is currently the only approved iron chelator for use in NTDT.[29,32]
Modulation of HbF
Hydroxyurea has been shown to be safe and effective in reducing transfusion requirements in patients with TI. While HbF inducers in general appear promising, larger randomised and controlled trials are needed before their use in TI is widely advocated.[33]
Stem cell transplantation
Stem cell transplantation is rarely performed in TI, and is reserved for those patients who develop transfusion dependency.[34,35]
Novel therapies
Many targeted therapies (discussed above) show promise in the future management of TI, and may be considered for use where it is cost-effective.'28-
Thalassaemia minor
TM is usually asymptomatic and characterised by a borderline or mild anaemia and thalassaemic red cell indices (microcytosis, hypochromia, elevated red cell count and Mentzer index (MCV/RCC ratio) <13. It must be differentiated from iron deficiency anaemia (IDA).
β-thalassaemia minor
The HbA2 level is typically raised, but may be masked by co-existing IDA (as IDA decreases the HbA2 level). In such individuals, the IDA should be treated prior to Hb electrophoresis.
Premarital and prenatal counselling are important in ß-thalassaemia minor. If the partner also has ß-thalassaemia minor, there is a 25% chance of a child born having MT.
No specific treatment is required for individuals with isolated ß-thalassaemia minor.
Alpha thalassaemia minor
Alpha thalassaemia minor (deletion or inactivation of 1 or 2 α globin genes) is usually asymptomatic. In contrast to ß-thalassaemia minor, the HbA2 level is normal or borderline low, and the diagnosis is suspected upon exclusion of other causes of a microcytic anaemia. DNA analysis may be employed in instances where confirmation is necessary, such as in prenatal testing. No specific treatment is required for individuals with isolated α-thalassaemia minor.
Declaration. None.
Acknowledgements. None.
Author contributions. Equal contributions.
Funding. None.
Conflicts of interest. None.
References
1. Alli NA, Patel M, Poole J, Goga Y, Krause A. Thalassaemia (part 1). S Afr Med J 2021;111(6):529-534. https://doi.org/10.7196/SAMJ.2021.v111i6.15724 [ Links ]
2. Yardumian A, Tefler P, Darbyshire P. Standards for the clinical care of children and adults with thalassaemia in the UK. Public Health England, 2008. http://sct.screening.nhs.uk/standardsandguidelines (accessed 5 May 2014). [ Links ]
3. Cappellini MD, Cohen A, Porter J, et al. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT). 3rd ed. Nicosia, Cyprus: Thalassaemia International Federation, 2014. [ Links ]
4. Vichinsky E, Levine L, Bhatia S, et al. Standards of Care Guidelines for Thalassemia. Oakland, California: Children's Hospital & Research Center Oakland, 2012. http://hemonc.cho.org/thalassemia/documents/SOCGuidelines2012.pdf (accessed 5 May 2014). [ Links ]
5. Ho PJ, Tay L, Lindeman R, Catley D, Bowden K. Australian guidelines for the assessment of iron overload and iron chelation in transfusion dependent thalassaemia major, sickle cell disease and other congenital anaemias. Internal Med J 2011;41(7):516-24. https://doi.org/10.1111/j.1445-5994.2011.02527.x [ Links ]
6. Angellucci E, Barosi G, Camaschella C, et al. Italian Society of Haematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica 2008;93(5):741-752. https://doi.org/10.3324/haematol.12413 [ Links ]
7. Anaemia Institute for Research and Education and the Thalassemia Foundation of Canada. Guidelines for the Clinical Care of Patients with Thalassaemia in Canada. New York, Ontario: Thalassaemia Foundation of Canada, 2009. http://www.thalassemia.ca/wp-content/uploads/Thalassemia-Guidelines_LR.pdf (accessed 30 August 2021). [ Links ]
8. Musallam KM, Angastiniotis M, Eleftheriou A, et al. Cross-talk between available guidelines for the management of patients with beta-thalassemia major. Acta Haematologica 2013;130:64-73. https://doi.org/10.1159/000345734 [ Links ]
9. Shah FT, Sayani F, Trompeter S, et al. Challenges of blood transfusions in ß-thalassemia. Blood Rev 2019;37:100588. https://doi.org/10.1016/j.blre.2019.100588 [ Links ]
10. Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet 2018;391(10116):155-167. https://doi.org/10.1016/S0140-6736(17)31822-6 [ Links ]
11. Taher AT, Cappellini MD. How I manage medical complications of ß-thalassemia in adults. Blood 2018;132(17):1781-1791. https://doi.org/10.1182/blood-2018-06-818187 [ Links ]
12. Vichinsky E, Levine L, Bhatia S, et al. Standards of care guidelines for thalassaemia, 2012. Oakland: Children's Hospital and Research Centre, 2012. https://thalassemia.com/documents/SOCGuidelines2012.pdf (accessed 30 August 2021). [ Links ]
13. Centers for Disease Control and Prevention. CDC Recommended Adult Immunization Schedule. CDC, 2015. http://www.cdc.gov/vaccines/schedules/hcp/adult.html (accessed 30 May 2021). [ Links ]
14. Meiring S, Hussey G, Jeena P, et al. Recommendations for the use of meningococcal vaccines in South Africa. South Afr J Infect Dis 2017;32:82-86. [ Links ]
15. Cassinerio E, Baldini IM, Alameddine RS, et al. Pregnancy in patients with thalassemia major: A cohort study and conclusions for an adequate care management approach. Ann Hematol 2017;96(6):1015-1021. https://doi.org/10.1007/s00277-017-2979-9 [ Links ]
16. Farmakis D, Triposkiadis F, Lekakis J, et al. Heart failure in haemoglobinopathies: Pathophysiology, clinical phenotypes, and management. Eur J Heart Fail 2017;19(4):479-489. https://doi.org/10.1002/ejhf.708 [ Links ]
17. Lal A. Iron in health and disease: An update. Indian J Pediatr 2020;87(1):58-65. https://doi.org/10.1007/s12098-019-03054-8 [ Links ]
18. Piga A. Impact of bone disease and pain in thalassemia. Hematology Am Soc Hematol Educ Program 2017(1):272-277. https://doi.org/10.1182/asheducation-2017.L272 [ Links ]
19. Demosthenous C, Vlachaki E, Apostolou C, et al. Beta-thalassemia: Renal complications and mechanisms: A narrative review. Hematology 2019;24(1):426-438. https://doi.org/10.1080/16078454.2019.1599096 [ Links ]
20. Cappellini MD, Porter J, Origa R, et al. Sotatercept, a novel transforming growth factor b ligand trap, improves anemia in ß-thalassemia: A phase II, open-label, dose-finding study. Haematologica 2019;104(3):477-484. https://doi.org/10.3324/haematol.2018.198887 [ Links ]
21. Motta I, Bou-Fakhredin R, Taher AT, et al. Beta thalassemia: New therapeutic options beyond transfusion and iron chelation. Drugs 2020;80:1053-1063. https://doi.org/10.1007/s40265-020-01341-9 [ Links ]
22. Cappellini MD, Porter JB, Vip Viprakasit V, et al. A paradigm shift on beta-thalassaemia treatment: How will we manage this old disease with new therapies. Blood Rev 2018;32:300-311. https://doi.org/10.1016/j.blre.2018.02.001 [ Links ]
23. Casu C, Presti VL, Oikonomidou PR, et al. Short-term administration of JAK2 inhibitors reduces splenomegaly in mouse models of beta-thalassemia intermedia and major. Haematologica 2018;103(2):e46-e9. [ Links ]
24. Taher AT, Karakas Z, Cassinerio E, et al. Blood 2018;131(2):263-265. https://doi.org/10.1182/blood-2017-06-790121 [ Links ]
25. Musallam KM, Rivella S, Vichinsky E, et al. Non-transfusion-dependent thalassemias. Haematologica 2013;98(6):833-844. https://doi.org/10.3324/haematol.2012.066845 [ Links ]
26. Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev 2012;26(Suppl 1):S3-S6. https://doi.org/10.1016/S0268-960X(12)70003-6 [ Links ]
27. Asadov C, Alimirzoeva Z, Mammadova T, et al. ß-Thalassemia intermedia: A comprehensive overview and novel approaches. Int J Hematol 2018;108:5-21. https://doi.org/10.1007/s12185-018-2411-9 [ Links ]
28. Ben Salah N, Bou-Fakhredin R, Mellouli F, et al. Revisiting beta thalassemia intermedia: Past, present, and future prospects. Hematology 2017;22(10):607-616. https://doi.org/10.1080/10245332.2017.1333246 [ Links ]
29. Taher A, Vichinsky E, Musallam K, et al. Guidelines for the Management of Non-Transfusion Dependent Thalassaemia (NTDT). Nicosia, Cyprus: Thalassaemia International Federation, 2013. [ Links ]
30. Taher A, El Rassi F, Isma'eel H, et al. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica 2008;93(10):1584-1586. https://doi.org/10.3324/haematol.13098 [ Links ]
31. Pakbaz Z, Fischer R, Fung E, et al. Serum ferritin underestimates liver iron concentration in transfusion independent thalassemia patients as compared to regularly transfused thalassemia and sickle cell patients. Pediatr Blood Cancer 2007;49(3):329-332. https://doi.org/10.1002/pbc.21275 [ Links ]
32. Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol 2013;92(11):1485-1493. https://doi.org/10.1007%2Fs00277-013-1808-z [ Links ]
33. Algiraigri AH, Wright NAM, Paolucci EO, et al. Hydroxyurea for nontransfusion-dependent β-thalassemia: A systematic review and meta-analysis. Hematol Oncol Stem Cell Ther 2017;10(3):116-125. https://doi.org/10.1016/j.hemonc.2017.02.002 [ Links ]
34. Borgna-Pignattti C. Modern treatment of thalassaemia intermedia. Brit J Haematol 2007;138:291-304. https://doi.org/10.1111/j.1365-2141.2007.06654.x [ Links ]
35. Taher A, Tyan PI. The spleen. In: MD Cappellini, A Cohen, J Porter, A Taher, V Viprakasit (eds). Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT). 3rd edition. Thalassaemia International Federation, 2014:126-133. [ Links ]
 Correspondence:
Correspondence:
N A Alli
nazeer.alli@nhls.ac.za
Accepted 18 June 2021


{kind=link}
{kind=link}
{kind=link}
{kind=link}