










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.110 n.12 Pretoria Dec. 2020
http://dx.doi.org/10.7196/samj.2020.v110i12.15328
REVIEW
Rapid evolution of our understanding of the pathogenesis of COVID-19 - implications for therapy
F MustafaI, II; R GilesIII; M S PepperIV
IMB ChB; Institute for Cellular and Molecular Medicine, Department of Immunology; and SAMRC Extramural Unit for Stem Cell Research and Therapy, Faculty of Health Sciences, University of Pretoria, South Africa
IIMB ChB; Department of Paediatrics, Faculty of Health Sciences, University of Pretoria, South Africa
IIIBSc, BSc Hons; Institute for Cellular and Molecular Medicine, Department of Immunology; and SAMRC Extramural Unit for Stem Cell Research and Therapy, Faculty of Health Sciences, University of Pretoria, South Africa
IVMB ChB, PhD, MD, PD; Institute for Cellular and Molecular Medicine, Department of Immunology; and SAMRC Extramural Unit for Stem Cell Research and Therapy, Faculty of Health Sciences, University of Pretoria, South Africa
ABSTRACT
COVID-19 severity appears to lie in its propensity to cause a hyperinflammatory response, attributed to the cytokine release syndrome (CRS) or 'cytokine storm', although the exact role of the CRS remains to be fully elucidated. Hyperinflammation triggers a hypercoagulable state, also thought to play a key role in COVID-19 pathogenesis. Disease severity is linked to age, sex and comorbid conditions, which in turn may be linked to oxidative stress and pre-existing depletion of nicotinamide adenine dinucleotide (NAD+). There is increasing evidence that the host genome may determine disease outcome. Since most information pertaining to COVID-19 has thus far been extrapolated from the 'global North', similar studies in African populations are warranted. Many studies are aimed at finding a therapeutic strategy based on scientific rationale. Some promising results have emerged, e.g. the use of corticosteroids in severe acute respiratory distress syndrome (ARDS).
The pandemic caused by the novel coronavirus SARS-CoV-2 manifests clinically in different ways. Most patients infected with SARS-CoV-2 exhibit mild to moderate symptoms and approximately 40 - 50% are asymptomatic. However, some patients develop severe disease that manifests as the acute respiratory distress syndrome (ARDS), multi-organ failure and/or septic shock. The virus is transmitted via aerosol, fomite and possibly faecal-oral routes.[1]
Most individuals who are admitted to hospital present with pneumonia and hypoxaemia.[1] Some individuals develop coagulation-related pathologies and multiple-organ failure, which sets COVID-19 apart from hyperinflammatory states seen in other diseases.[2]
Methods
PubMed and other databases were accessed using the search words 'COVID-19 AND pathophysiology', and 'COVID-19 AND treatment AND therapeutic strategies'. The authors then evaluated published studies on the pathophysiology, severity and therapeutic options based on data from these sources. This is not an exhaustive systematic review, or a meta-analysis, but rather a descriptive review, in an ever-changing field, of some of the key pathogenetic mechanisms to have emerged in a disease whose existence we have known about for less than a year.
Pathogenesis of COVID-19
SARS-CoV-2 infects the host by binding to angiotensin-converting enzyme 2 (ACE-2). There is now evidence that the virus infects multiple pulmonary cell types, including bronchial epithelium, with the virus being detected in abundance in ciliated cells, non-ciliated mucus-secreting (goblet) cells, club cells and alveolar cells.[3-5] The receptor is also found in the kidney, heart and on gut cells.[6] SARS-CoV-2 also targets the immune system, where nucleocapsid proteins target B cells and small blood vessels which is thought to contribute to systemic vasculitis and immune dysfunction.[4] Other receptors on human cells likely to promote entry of SARS-CoV-2 include transmembrane serine protease 2,[7] sialic acid[8] and extracellular matrix metalloproteinase inducer CD147,[9] which are present in vascular endothelial cells, arterial smooth-muscle cells and other cell types. Descriptive studies on the presence of ACE-2 and other (co)receptor proteins and/or transcripts[10] imply that they may be necessary but not that they are sufficient for viral binding. As a result of viral binding to ACE-2, concern has been expressed about using renin-angiotensin-aldosterone system inhibitors, such as angiotensin II receptor blockers (ARBs) and ACE inhibitors for the treatment of hypertension in COVID-19 patients.[11-13] However, two large studies have demonstrated that there is no evidence for an increased risk of COVID-19 as a result of ACE inhibitor or ARB treatment.[14,15] The American Heart Association has stated that 'Patients taking ACE-i and ARBs who contract COVID-19 should continue treatment, unless otherwise advised by their physician.'[16]
The hyperinflammatory state that characterises COVID-19 appears to be linked in part to endothelial dysfunction which may result from viral binding to endothelial and pulmonary vascular smooth-muscle cell ACE-2 receptors.[3-5,10] Recent evidence has shown that interleukin (IL)-6, which plays an important role in the development and progression of the inflammatory process during viral infection, can be induced by angiotensin II through a mineralocorticoid receptor-dependent mechanism, contributing to the hyperinflammatory state.[17]
Immune dysregulation
Following infection, the host attempts to eliminate the virus by inducing an antiviral immune response, which includes both innate and adaptive immune systems.[18] The innate immune response is invoked at the first encounter with the virus and provides an initial mechanism for eliminating the pathogen. However, if the response is defective, the virus will proliferate and cause widespread damage to affected tissues.[19] Infected tissues undergo pyroptosis, which results in the release of damaged associated molecular patterns (DAMPs). These molecular patterns are recognised as 'foreign' by epithelial cells, endothelial cells and alveolar macrophages, which results in an uncontrolled innate inflammatory response in the lungs. This is mediated by pro-inflammatory cytokines.[18-20]
One of the consequences of SARS-CoV2 infection is the activation of neutrophils that release neutrophil extracellular traps (NETs).[21] NETs are comprised of nuclear and mitochondrial DNA and protein material from nuclear chromatin and cytosolic granules. One variant of NET, termed suicidal NETosis, results in cell death and the release of reactive oxygen species. Various molecules can promote this type of NET, including DAMPs. Damaged neutrophils have a negative redox potential that maintains endogenous self-antigens, such as high-mobility group box 1 (HMGB1) in a fully reduced form (fr-HMGBl). HMGB1 promotes activation of the receptor for advanced glycation end products (RAGE), which further triggers neutrophil-mediated inflammation.[21] Activation of toll-like receptor 4 (TLR-4) in turn acts as a procoagulant factor through platelet stimulation of this receptor.
The induction of a proinflammatory process is reflected in raised plasma concentrations of a number of cytokines and chemokines which include IL-1 beta (IL-1P), IL-1RA, IL-6, IL-7, IL-8, IL-9, IL-10, fibroblast growth factor (FGF), granulocyte colony stimulating factor (G-CSF), granulocyte macrophage colony stimulating factor (GM-CSF), interferon-gamma (IFN-y), IFN-y inducible protein (IP)-10, monocyte chemotactic protein (MCP)-1, monocyte inhibitory protein (MIP)-IA, MIP-1B, platelet-derived growth factor (PDGF), tumour necrosis factor-alpha (TNF-a), and vascular endothelial growth factor (VEGF).[3,18,22-24] These cytokines and chemokines drive inflammation. In mild diseases with limited inflammation, this pro-inflammatory state results in the production of neutralising antibodies by plasma cells driven by virus-specific T cells, and eventually progresses to eradication of the virus and recovery. However, in severe COVID-19 there is an exaggerated degree of inflammation which is deleterious to the host.
In COVID-19, the accumulation of pro-inflammatory cytokines may result in the cytokine release syndrome (CRS or 'cytokine storm') with systemic inflammation and damage to the respiratory infrastructure, as well as a hypercoagulable state as a consequence of endothelial injury. In patients with more severe disease, higher levels of IL-2, IL-6, IL-7 IL-10, G-CSF, IP-10, MCP-1, MIP-1A and TNF-a have been observed compared with patients with milder infection and those who do not require admission to an intensive care unit (ICU)[25]
Recent evidence indicates that the surge of cytokines thought to be the driver of the CRS in COVID-19 may not be as high as is seen in other causes of ARDS, and the CRS itself lacks a clear definition with some authors denying its existence. In essence, the presence of immune mediators such as chemokines, interferons, TNF-a, interleukins and others is part of a fundamental immune response needed for innate infection control. It has been proposed that in the CRS, these immune mediators are raised to levels that are detrimental to the host. Of particular interest is IL-6, a key mediator of acute inflammation, which has gained much attention over the course of the pandemic. IL-6 has become a therapeutic target for COVID-19. In randomised clinical trials conducted by the National Heart, Lung and Blood Institution's ARDS Network, it was reported that patients with severe disease who develop ARDS due to COVID-19 have IL-6 levels 10 - 40-times higher than the upper limit of normal.[26] The phenotype associated with typical ARDS manifests as elevated plasma levels of pro-inflammatory cytokines, an increased propensity to shock and poor clinical outcomes. While the phenotype in COVID-19-related ARDS is consistent with what is expected in a cytokine storm, IL-6 levels in ARDS due to other causes are 10 - 200 times higher than those seen in ARDS due to COVID-19, casting doubt on the presence of a CRS in COVID-19.[26] There is a need to identify patients in whom targeted immunotherapeutic monoclonal antibodies are effective, and to determine biomarkers that could potentially predict the response, to further understand the CRS in COVID-19.[26]
Evidence for macrophage involvement in the pathogenesis of SARS-CoV-2 is undisputed, and it has been suggested that macrophages in the lungs and other organs in COVID-19 are newly derived from circulating monocytes rather than specialised local pre-existing resident tissue macrophages. Circulating monocytes exit the bone marrow via the CC chemokine receptor 2 (CCR2). Trials targeting the chemokine receptors (involved in regulation of monocytes and in T-cell migration) are ongoing.[2]
Coronaviruses are different from other respiratory viruses in that they induce interferons. INF-a and INF-P can drive antiviral immunity, but dysregulated release leads to extensive pathology as seen in patients with severe COVID-19. Normally, interferons mainly have antiviral activity, activating natural killer (NK) cells and macrophages and upregulating the expression of major histocompatibility complex (MHC) antigens which facilitate binding of the virus to T cells. Whereas INF-a and INF-P are expressed almost everywhere, INF-X affects mostly epithelial cells and has an effect on T-helper type 2 (TH2) cells; it is also immunosuppressive. As a therapeutic intervention, blocking this cytokine might potentially prevent the CRS.[27] The unchecked production of INF-y also leads to dysregulated macrophage activation. Clinical trials assessing the effects of INF-y blockade in patients with respiratory distress and severe disease are under way.[27]
Host factors promoting COVID-19 severity
Age
Advanced age (>60 years) and certain comorbidities are presumed to contribute to a dysregulated immune response, which is less efficient at pathogen elimination.[28] This process describes immunosenescence and has been attributed to altered dendritic cell maturation and defective T-cell activation. With increased age, the combination of thymic degeneration and the continuous conversion of naïve T cells into memory T cells as a result of antigen challenge results in the T-cell receptor repertoire being reduced and replaced by oligoclonal memory T cells.[29] Age-related alterations in the T-cell repertoire have significant clinical implications, since naïve T cells have mostly been replaced. This is of particular concern in the production of a vaccine for COVID-19 if one draws a parallel with the poor outcomes seen with the yearly influenza vaccine among the elderly.[29] Ageing is considered to be a pro-inflammatory state. Taken together, changes seen with advanced age including a dysregulated immune response and an augmented inflammatory state can exacerbate disease severity.[18] A recent study has shed light on the effects of age on pulmonary defence mechanisms: specific factors in the antioxidant defence system of alveolar type II cells, including superoxide dismutase 3 and activating transcription factor 4 (an endoplasmic reticulum stress sensor), become less effective with age.[30]
In contrast to those of advanced age, children are less severely affected despite having higher viral titres; 50% of individuals less than 18 years of age experience mild disease or are asymptomatic, and less than 6% develop severe symptomatic infection. Mortality in this group is extremely low in comparison with adults and especially the elderly.[31]
Comorbidities
Comorbidities appear to be the most consistent feature in individuals with severe disease, with more than 30% of symptomatic individuals having a comorbid condition. The vast majority of severe COVID-19 cases and deaths occur in individuals with an underlying chronic condition including obesity, diabetes, chronic respiratory disease, diseases of the cardiovascular system (especially hypertension) and malignancy.[18] These conditions are all associated with an impaired immune response, and when superimposed on a hyperinflammatory state, contribute to disease severity. These comorbidities are also associated with endothelial dysfunction.[32]
In addition, there is increasing evidence that a deficiency in nicotinamide adenine dinucleotide (NAD+) is associated with disease severity in COVID-19. Hypertension, obesity, diabetes and advanced age are all associated with oxidative stress and pre-existing depletion of NAD+. When patients with comorbidities are infected with SARS-CoV-2, this reduces NAD+ dependent silent information regulator 1 (SIRT1) production.[33] SIRT1 controls and modifies the inflammatory response and, together with other members of the sirtuin family, constitutes part of the primary defence against DNA and RNA viral pathogens. Nutritional support with NAD+ precursors and SIRT1 activators (including zinc and nicotinic acid) has a rational scientific basis.[33]
Genetic factors
HLA haplotypes and SARS-CoV-2 infection[19]
Human leukocyte antigen (HLA) diversity is a product of selective pressure during pathogen co-evolution. Genetic susceptibility to various infectious diseases such as tuberculosis, leprosy, HIV, hepatitis B and influenza has been well established. This susceptibility is conferred in part by HLA loci. For example, HLA-A*11, HLA-B*35, and HLA-DRB1*10 confer susceptibility to influenza A(H1N1)pdm09 infection. Hence it is important to identify possible HLA haplotypes which may be associated with an immune response to SARS-CoV-2. This information will be beneficial for vaccine development. It may also help in developing novel treatment strategies.
ARDS
Complex genetic and environmental risk factors contribute to the development of ARDS. More than 80 genes have been implicated, with the most common variants being found in the genes for IL-6, IL-10, ACE, IL-1 receptor antagonist (IL1RN), mannose-binding lectin 2 (MBL-2), nicotinamide phosphoribosyltransferase (NAMPT) and VEGF-A. These gene products play an important role in the response to external stimuli and in signal transduction, as well as in the immune response, chemotaxis and cell proliferation.[34] Since most of this information has been extrapolated from European populations, similar studies in African populations (which have extensive genomic diversity) would be important.
Prior to COVID-19 and in other diseases with ARDS, in a genome-wide association study Christie et al.[35] identified variants associated with ARDS in the genes for tyrosine phosphatase receptor type f polypeptide interacting protein (liprin) and alpha 1 (PPF1A1), as well as genes encoding IL-10, angiopoietin 2 (ANGPT2), Fas cell surface death receptor (FAS) and myosin light chain kinase (MYLK). Lee et al.[36] identified MYLK to be associated with ARDS and 'ventilator free days', linking MYLK to ARDS susceptibility. Shortt et al.[37] presented three genomic variants that may be involved in enhancing the susceptibility, severity and clinical outcome of ARDS that include arylsulfatase D gene (ARSD), X Kell blood group complex subunit related family member 3 gene (XKR3) and zinc finger protein 335 (ZNF335).
COVID-19
A genome-wide analysis study (GWAS) in Italian and Spanish patients with confirmed SARS-CoV-2 found that loci on chromosomes 3 and 9 were linked to SARS-CoV-2-induced respiratory failure.[38] These loci include: rs11385942 insertion-deletion GA or G variant at locus 3p21.31 and rs657152 A or C SNP at locus 9q34.2.[38] The association signal at locus 3p21.31 consists of 6 genes that include CCR9, CXCR6, FYCO1, LZTFL1, SLC6A20 and XCR1. Rs11385942 contains the risk allele GA and has been linked to the reduced expression of CXCR6 and increased expression of SLC6A20 and LZTFL1, which is highly expressed in pulmonary bronchial and alveolar cells in humans.[38]
When patients on mechanical ventilation were compared with those on supplemental oxygenation, the former were more likely to have the 3p21.31 (rs11385942) risk allele.[38] This suggests that the 3p21.31 locus could be involved in enhanced susceptibility to more severe disease. This study also showed that patients with blood group A were found to be at a higher risk of developing SARS-CoV-2 compared with other blood groups, while blood group O had a protective effect when compared with other blood groups. Two subsequent studies based on clinical data and outcomes, but which did not involve a GWAS, yielded similar outcomes. Hoilend et al.[39]reported that patients with blood group A or AB had an increased risk for requiring mechanical ventilation, continual renal replacement therapy and prolonged ICU stay when compared with patients with O and B blood groups, while Barnkob et al.[40]reported that ABO blood groups could define susceptibility to infection (with O blood group being protective) but not hospitalisation or death from COVID-19. However, the possible role of blood groups in determining COVID-19 susceptibility and severity has been called into question.[41]
Other contributors to disease severity and complications of SARS-CoV-2 infection
The presence of ACE-2 receptors on endothelial cells, pericytes and smooth-muscle cells in most organs suggests that SARS-CoV-2, once present in the circulation, may easily spread to all body systems. A recent study of postmortem lung tissues from patients who died from COVID-19 and influenza A (H1N1) found a significant number of ACE-2-positive endothelial cells and significant morphological changes with disruption of intercellular junctions, cell swelling, and a loss of contact with the basement membrane.[42] These findings suggest that endothelial dysfunction, including systemic vasculitis and endothelial cell apoptosis, contributes to the increased risk of developing venous thromboembolic disease and inflammation in various organs in COVID-19.[43-45] It should be borne in mind, as indicated above, that ACE-2 may be necessary but not sufficient for SARS-CoV-2 entry; caution should therefore be employed in interpreting localisation data.
It is also probable that, as with other infectious diseases, SARS-CoV-2 can activate coagulopathy through inflammatory responses. Platelet-dense granules contain polyphosphates which are secreted upon activation.[46] Polyphosphates released from activated platelets accelerate factor V activation, inhibit the anticoagulant activity of tissue factor pathway inhibitor, promote factor XI activation by thrombin, and contribute to the synthesis of thicker fibrin strands that are resistant to fibrinolysis.[47] This suggests a link between the immune system and the coagulation cascade in COVID-19, as with all sepsis aetiologies.
In addition, a new mechanism has been described in which ACE-2 receptor-bound SARS-CoV-2 induces luminal expression of tissue factor (TF). TF then binds to circulating coagulation factor VII, resulting in production of excess thrombin and fibrin and increased intrinsic clot formation.[48] A number of cytokines (primarily TNF-a) upregulate NAD phosphate (NADPH) oxidase and nuclear factorkappa B (NF-k B) to induce TF production in endothelial cells.[48]
Inflammation also results in activated pulmonary vascular endothelial cells and endothelial injury, with a resultant prothrombotic state in the pulmonary vasculature.[49] Vascular endothelial injury causes further thrombocytopenia (through peripheral platelet destruction), a reduction in natural anticoagulants and haemostatic activation, and manifests as diffuse intravascular coagulation in COVID-19.[49]
Multisystem inflammatory syndrome in children
A rare but severe form of COVID-19 has been reported in children and is referred to as the multisystem inflammatory syndrome in children (MIS-C).[50] Clinical features of MIS-C are reported to be similar to but distinct from Kawasaki disease.
MIS-C is not an adult version of the CRS. Ongoing studies suggest that antibodies to SARS-CoV-2 actually permit viral entry into affected cells[51] or that immune complexes may be stimulatory to the immune system.[51] Cross-reactivity between a microbial antigen and host tissues, where epitopes are shared (molecular mimicry), is also a possibility.[52] Lastly, it is possible that SARS-CoV-2 may act as an adjuvant factor for the, as yet, unknown trigger in MIS-C.[50]
Implications for therapy
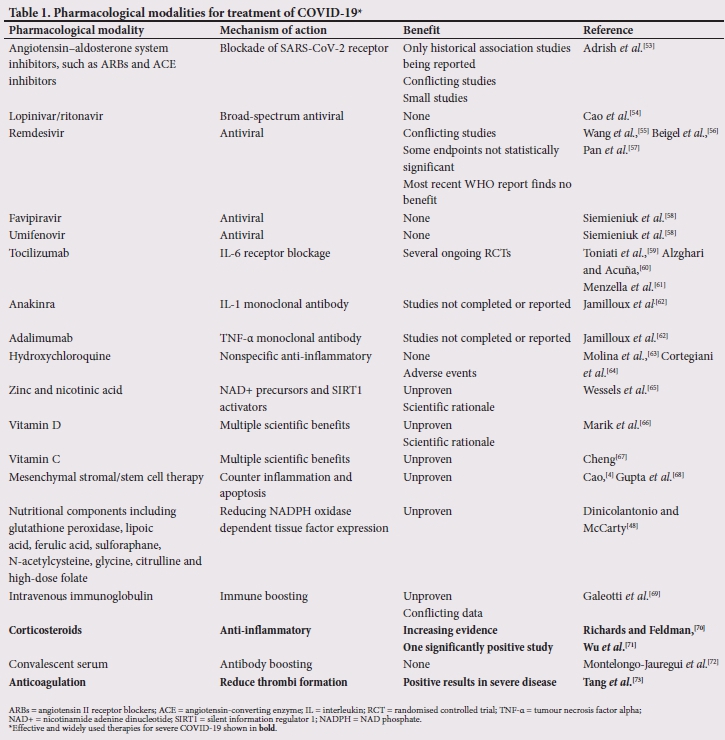
Multiple therapeutic strategies are being considered for patients with more severe COVID-19 and these are summarised in Table 1.[4,48,53-73] Some (in bold in the table) are effective and widely used therapies for severe COVID-19. The jury is still out as to the value of antiviral therapies. Recent trials using lopinivar/ritonavir as a single agent have shown no benefit.[54] A variety of antiviral agents are being studied in various clinical trials.[74] A study published in The Lancet reported that remdesivir was not associated with a difference in time to clinical improvement (hazard ratio 1.23; 95% confidence interval (CI) 0.87 -1.75).[55] In a subsequent study, in patients with symptom duration of 10 days or less, those treated with remdesivir had a numerically faster time to clinical improvement compared with those receiving placebo (hazard ratio 1.52; 95% CI 0.95 - 2.43).[56]
Several clinical trials have assessed the efficacy of hydroxy-chloroquine (alone or in combination) based on its nonspecific anti-inflammatory effects seen in other conditions. Data generated thus far indicate that it lacks efficacy and there may be safety concerns related to its use.[63]
Trials are under way to assess the potential benefits of therapies that block IL-6, its receptor, IL-1β and GM-CSF, as well as other myeloid-derived inflammatory cytokines, especially TNF-a. IL-1 is a crucial element in the development of the CRS in patients with severe disease. Anakinra (an IL-1 antagonist) has been shown to be of benefit in patients with severe sepsis complicated by macrophage activation syndrome (MAS); studies with anakinra in COVID-19 may be useful to further understand the disease process, as well as providing a potential therapeutic option.[25] TNF-a is one of the major drivers of inflammatory responses. TNF-a antagonistic drugs are used widely in management of rheumatological diseases. TNF-a levels are moderately elevated in SARS-CoV-2 patients. Targeting TNF-a has thus been identified as a potential therapeutic option, and clinical trials have been initiated to assess the effect of adalimumab, an anti-TNF monoclonal antibody.[24] Strategies to interfere with receptor binding and cytokine signalling may unveil crucial pathogenetic mechanisms and possibly therapeutic options to prevent severe disease.
It is possible that high-dose intravenous immunoglobulin (IVIg) may have a beneficial effect in the hyperinflammatory phase but the mechanism of action is not known.[69,75] It is particularly widely used in children with MIS-C.
Several nutritional components may have theoretical, but as yet untested, benefits in reducing NADPH oxidase dependent TF expression, including glutathione peroxidase, lipoic acid, ferulic acid, sulforaphane, N-acetylcysteine, glycine, citrulline and high-dose folate.[48] Studies are planned or are under way to identify nutritional supplements which may help with NAPDH reduction as part of the treatment regimen in COVID-19.[48]
The use of corticosteroids for the treatment of COVID-19, based on their ability to suppress an exaggerated inflammatory response, has been widely deliberated in medical literature. Currently corticosteroid use for treatment of COVID-19 is recommended, mostly based on evidence from the RECOVERY trial (Randomized Evaluation of COVID Therapy).[76] The RECOVERY trial reported that the use of corticosteroids (dexamethasone and methylprednisone) reduced the risk of mortality, the need for mechanical ventilation and duration of hospital stay.[76] It is important to note that corticosteroids have shown no benefit in patients who do not require respiratory support[70] The Surviving Sepsis Guideline (SSG) for COVID-19 recommends the use of low-dose corticosteroids in COVID-19 patients with shock who are not responding to treatment and in patients requiring mechanical ventilation, while it advises against steroid use for patients with COVID-19 in respiratory failure but without ARDS.[77] Studies from China propose the use of corticosteroids early in COVID-19 based on the observation of reduced disease escalation and improved outcomes.[71]
Randomised clinical trials on the treatment of children with MIS-C have not been reported but certain therapies are widely used, including systemic steroids, high-dose IVIg and cytokine inhibitors. In addition, patients require respiratory and inotropic support.[78]
Conclusions
The disease course of COVID-19, including its molecular and mechanistic components, is becoming clearer. Three distinct stages of clinical progression based on immunological parameters have been described. The first involves an antiviral response with an immediate potent interferon release which brings the infection under control and potentially eradicates the virus. During the second stage, a delayed interferon response may ensue which would nonetheless result in extensive damage. If the second stage fails to achieve infection resolution, there is progression to a more severe third stage in which hyperinflammation with dysregulated macrophage activation and widespread coagulation ensues. Patients who progress to stage three may also have dysregulated repair mechanisms and fibrosis.[2] The systemic hyperinflammation seen in patients infected with SARS-CoV-2 is believed by many to be the nidus of the pathology in severe disease, although a clearer definition of the CRS, particularly in relation to other causes, is needed. It is critical that the pathways involved in these stages are understood in order to develop appropriate treatment strategies.
Declaration. None.
Acknowledgements. None.
Author contributions. The authors contributed equally to writing the manuscript, and have read and agreed to the published version.
Funding. This work was supported by grants from the South African Medical Research Council Extramural Unit for Stem Cell Research and Therapy, and the Institute for Cellular and Molecular Medicine of the University of Pretoria (MSP).
Conflicts of interest. None.
References
1. Mackenzie JS, Smith DW. Covid-19: A novel zoonotic disease caused by a coronavirus from China: What we know and what we don't. Microbiol Aust 2020;41(1):45-50. https://doi.org/10.1071/MA20013 [ Links ]
2. Merad M, Martin JC. Pathological inflammation in patients with Covid-19: A key role for monocytes and macrophages. Nature Rev Immunol 2020:20(6):355-362. https://doi.org/10.1038/s41577-020-0331-4 [ Links ]
3. Huertas A, Montani D, Savale L, et al. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur Respir J 2020;56:2001634. https://doi.org/10.1183/13993003.01634-2020 [ Links ]
4. Cao X. Covid-19: Immunopathology and its implications for therapy. Nature Rev Immunol 2020;20(5):269-270. https://doi.org/10.1038/s41577-020-0308-3 [ Links ]
5. Pice LC, McCabe C, Garfield B, et al. Thrombosis and COVID-19 pneumonia: The clot thickens! Eur Respir J 2020;56:2001608. https://doi.org/10.1183/13993003.01608-2020 [ Links ]
6. Gheblawi M, Wang K, Viveiros A, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ACE2. Circ Res 2020;126(10):1456-1474. https://doi.org/10.1161/CIRCRESAHA.120.317015 [ Links ]
7. Matsuyama S, Nao N, Shirato K, et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci USA 2020;117(13):7001-7003. https://doi.org/10.1073/pnas.2002589117 [ Links ]
8. Tortorici MA, Walls AC, Lang Y, et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat Struct Mol Biol 2019;26:481-489. https://doi.org/10.1038/s41594-019-0233-y [ Links ]
9. Chen Z, Mi L, Xu J, et al. Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J Infect Dis 2005;191(5):755-760. https://doi.org/10.1086/427811 [ Links ]
10. Hamming I, Timens W, Bulthuis ML, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus: A first step in understanding SARS pathogenesis. J Pathol 2004;203(2):631-637. https://doi.org/10.1002/path.1570 [ Links ]
11. Guo J, Huang Z, Lin L, et al. Coronavirus disease 2019 (COVID-19) and cardiovascular disease: A viewpoint on the potential influence of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 infection. J Am Heart Assoc 2020;9(7):e016219. https://doi.org/10.1161/JAHA.120.016219 [ Links ]
12. Mourad JJ, Levy BI. Interaction between RAAS inhibitors and ACE2 in the context of COVID-19. Nat Rev Cardiol 2020;17:313. https://doi.org/10.1038/s41569-020-0368-x [ Links ]
13. Vaduganathan M, Vardeny O, Michel T, et al. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med 2020;382:1653-1659. https://doi.org/10.1056/NEJMsr2005760 [ Links ]
14. Mancia G, Rea F, Ludergnani M, et al. Renin-angiotensin-aldosterone system blockers and the risk of Covid-19. N Engl J Med 2020;382:2431-2440. https://doi.org/10.1056/NEJMoa2006923 [ Links ]
15. Reynolds HR, Adhikari S, Pulgarin C, et al. Renin-angiotensin-aldosterone system inhibitors and risk of Covid-19. N Engl J Med 2020;382:2441-2448. https://doi.org/10.1056/NEJMoa2008975 [ Links ]
16. American Heart Association. Patients taking ACE-i and ARBs who contract COVID-19 should continue treatment, unless otherwise advised by their physician. 17 March 2020. https://newsroom.heart.org/news/patients-taking-ace-i-and-arbs-who-contract-covid-19-should-continue-treatment-unless-otherwise-advised-by-their-physician (accessed 14 September 2020). [ Links ]
17. Luther JM, Gainer JV, Murphey LJ, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 2006;48(6):1050-1057. https://doi.org/10.1161/01.HYP.0000248135.97380.76 [ Links ]
18. Saghazadeh A, Rezaei N. Immune-epidemiological parameters ofthe novel coronavirus -a perspective. Expert Rev Clin Immunol 2020:16(5):465-470. https://doi.org/10.1080/1744666X.2020.1750954 [ Links ]
19. Shi Y, Wang Y, Shao C, et al. Covid-19 infection: The perspectives on immune responses. Cell Death Differ 2020;27(5):1451-1454. https://doi.org/10.1038/s41418-020-0530-3 [ Links ]
20. Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with covid-19 in Wuhan, China. Clin Infect Dis 2020 Mar 12;ciaa248(epub ahead of print). https://doi.org/10.1093/cid/ciaa248 [ Links ]
21. Cicco S, Cicco G, Racanelli V, Vacca A. Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): Two potential targets for COVID-19 treatment. Mediators Inflamm 2020;2020:7527953(epub ahead of print). https://doi.org/10.1155/2020/7527953 [ Links ]
22. Feldmann M, Maini RN, Woody JN, et al. Trials of anti-tumour necrosis factor therapy for covid-19 are urgently needed. Lancet 2020; 395(10234):1407-1409. https://doi.org/10.1016/S0140-6736(20)30858-8 [ Links ]
23. Yang Y, Shen C, Li J, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol 2020;146(1):119-127.e4. https://doi.org/10.1016/j.jaci.2020.04.027 [ Links ]
24. Sarzi-Puttini P, Giorgi V, Sirotti S, et al. Covid-19, cytokines and immunosuppression: What can we learn from severe acute respiratory syndrome? Clin Exp Rheumatol 2020;38(2):337-342. PMID: 32202240 [ Links ]
25. Mehta P, McAuley DF, Brown M, et al. Covid-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020;395(10229):1033. https://doi.org/10.1016/S0140-6736(20)30628-0 [ Links ]
26. Sinha P, Matthay MA, Calfee CS. Is a 'cytokine storm' relevant to COVID 19? JAMA Intern Med 2020;180(9):1152-1154(epub ahead of print 30 June 2020). https://doi.org/10.1001/jamainternmed.2020.3313 [ Links ]
27. Russell B, Moss C, George G, et al. Associations between immune-suppressive and stimulating drugs and novel COVID-19 - a systematic review of current evidence. ecancermedicalscience 2020;14:1022. https://doi.org/10.3332/ecancer.2020.1022 [ Links ]
28. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LF. The trinity of Covid-19: Immunity, inflammation and intervention. Nature Rev Immunol 2020:20(6):363-374. https://doi.org/10.1038/s41577-020-0311-8 [ Links ]
29. Vallejo AN. Age-dependent alterations of the T cell repertoire and functional diversity of T cells of the aged. Immunologic Res 2006;36(1-3):221-228. https://doi.org/10.1385/IR:36:1:221 [ Links ]
30. Abouhashem AS, Singh K, Azzazy HM, et al. Is low alveolar type II cell SOD3 in the lungs of elderly linked to the observed severity of COVID-19? Antioxid Redox Signal 2020;33:59-65. https://doi.org/10.1089/ars.2020.8111 [ Links ]
31. Bialek S, Gierke R, Hughes M, McNamara LA, Pilishvili T, Skoff T. Coronavirus disease 2019 in children - United States, February 12 - April 2, 2020. MMWR Morb Mortal Wkly Rep 2020;69(14):422-426. https://doi.org/10.15585/mmwr.mm6914e4 [ Links ]
32. Huertas A, Guignabert C, Barbera JA, et al. Pulmonary vascular endothelium: The orchestra conductor in respiratory diseases: Highlights from basic research to therapy. Eur Respir J 2018;51(4):1700745. https://doi.org/10.1183/13993003.00745-2017 [ Links ]
33. Miller R, Wentzel AR, Richards GA. COVID-19: NAD+ deficiency may predispose the aged, obese and type 2 diabetics to mortality through its effect on SIRT1 activity. Med Hypotheses 2020;144:110044. https://doi.org/10.1016/j.mehy.2020.110044 [ Links ]
34. Hernández-Beeftink T, Guillen-Guio B, Villar J, Flores C. Genomics and the acute respiratory distress syndrome: Current and future directions. Int J Mol Sci 2019;20(16):4004. https://doi.org/10.3390/ijms20164004 [ Links ]
35. Christie JD, Wurfel MM, Feng R, et al. Genome wide association identifies PPFIA1 as a candidate gene for acute lung injury risk following major trauma. PLoS ONE 2012;7(1):e28268. https://doi.org/10.1371/journalpone.0028268 [ Links ]
36. Lee S, Emond MJ, Bamshad MJ, et al Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Human Genetics 2012; 91(2):224-37. https://doi.org/10.1016/j.ajhg.2012.06.007 [ Links ]
37. Shortt K, Chaudhary S, Grigoryev D, et al. Identification of novel single nucleotide polymorphisms associated with acute respiratory distress syndrome by exome-seq. PLoS ONE 2014;9(11):e111953. https://doi.org/10.1371/journalpone.0111953 [ Links ]
38. Ellinghaus D, Degenhardt F, Bujanda L, et al. Genomewide association study of severe Covid-19 with respiratory failure. N Engl J Med 2020;383(16):1522-1534. https://doi.org/10.1056/NEJMoa2020283 [ Links ]
39. Holland RL, Fergusson NA, Mitra AR, et al. The association of ABO blood group with indices of disease severity and multiorgan dysfunction in COVID-19. Blood Adv 2020;4:4981-4989. https://doi.org/10.1182/bloodadvances.2020002623 [ Links ]
40. Barnkob MB, Pottegard A, Stovring H, et al. Reduced prevalence of SARS-CoV-2 infection in ABO blood group O. Blood Adv 2020;4:4990-4993. https://doi.org/10.1182/bloodadvances.2020002657 [ Links ]
41. Latz CA, DeCarlo C, Boitano L, et al. Blood type and outcomes in patients with COVID-19. Ann Hematol 2020;Jul 12:1-6(epub ahead of print). https://doi.org/10.1007/s00277-020-04169-1 [ Links ]
42. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 2020;383(2):120-128. https://doi.org/10.1056/NEJMoa2015432 [ Links ]
43. Criel M, Falter M, Jaeken J, et al. Venous thromboembolism in SARS-CoV-2 patients: Only a problem in ventilated ICU patients, or is there more to it? Eur Respir J 2020;56:2001201. https://doi.org/10.1183/13993003.01201-2020 [ Links ]
44. Poissy J, Goutay J, Caplan M, et al Pulmonary embolism in COVID-19 patients: Awareness of an increased prevalence. Circulation 2020;142(2):184-186. https://doi.org/10.1161/CIRCULATIONAHA.120.047430 [ Links ]
45. Ullah W, Saeed R, Sarwar U, et al. COVID-19 complicated by acute pulmonary embolism and right-sided heart failure. JACC Case Rep 2020;2(9):1379-1382. https://doi.org/10.1016Zj.jaccas.2020.04.008 [ Links ]
46. Ruiz FA, Lea CR, Oldfield E, et al. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem 2004;279:44250-44257. https://doi.org/10.1074/jbc.M406261200 [ Links ]
47. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 2013;13:34-45. https://doi.org/10.1038/nri3345 [ Links ]
48. Dinicolantonio JJ, McCarty M. Thrombotic complications of COVID-19 may reflect an upregulation of endothelial tissue factor expression that is contingent on activation of endosomal NADPH oxidase. Open Heart 2020;7e001337. https://doi.org/10.1136/openhrt-2020-001337 [ Links ]
49. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020;135(23):2033-2040. https://doi.org/10.1182/blood.2020006000 [ Links ]
50. Hendricks C, Mustafa F, Green RJ, Pepper MS. Paediatric multisystem inflammatory syndrome. S Afr Med J 2020;110(9):832-834. https://doi.org/10.7196/SAMJ.2020.v110i9.15062 [ Links ]
51. Whittaker E, Bamford A, Kenny J, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA 2020;324(3):259. https://doi.org/10.1001/jama.2020.10369 [ Links ]
52. Raba AA, Abobaker A. Covid-19 and Kawasaki disease: An etiology or coincidental infection. Pediatr Infect Dis J 2020;39(8):213. https://doi.org/10.1097/INF.0000000000002779 [ Links ]
53. Adrish M, Chilimuri S, Sun H, Mantri N, Yugay A, Zahid M. The association of renin-angiotensin-aldosterone system inhibitors with outcomes among a predominantly ethnic minority patient population hospitalized with COVID-19: The Bronx experience. Cureus 2020;12(9):e10217. https://doi.org/10.7759/cureus.10217 [ Links ]
54. Cao B, Wang Y, Wen D, et al A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med 2020;382(19):1787-1799. https://doi.org/10.1056/NEJMoa2001282 [ Links ]
55. Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020;395(10236):1569-1578. https://doi.org/10.1016/S0140-6736(20)31022-9 [ Links ]
56. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19: Preliminary report. N Engl J Med 2020; 22 May(epub ahead of print) 2020 . https://doi.org/10.1056/NEJMoa2007764 [ Links ]
57. WHO Solidarity Trial Consortium, Pan H, Peto PR, Abdool Q, et al Repurposed antiviral drugs for COVID-19: Interim WHO SOLIDARITY trial results. medRxiv 2020.10.15.20209817.https://doi.org/10.1101/2020.10.15.20209817 [ Links ]
58. Siemieniuk RAC, Bartoszko JJ, Ge L, et al. Drug treatments for Covid-19: Living systematic review and network meta-analysis. BMJ 2020;370:m2980. https://doi.org/10.1136/bmj.m2980 [ Links ]
59. Toniati P, Piva S, Cattalini M, et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun Rev 2020;19(7):102568. https://doi.org/10.1016/j.autrev.2020.102568 [ Links ]
60. Alzghari SK, Acuna VS. Supportive treatment with tocilizumab for COVID-19: A systematic review. J Clin Virol 2020;127:104380. https://doi.org/10.1016/j.jcv.2020.104380 [ Links ]
61. Menzella F, Biava M, Barbieri C, Livrieri F, Facciolongo N. Drugs Context 2020;9:2020-4-6. https://doi.org/10.7573/dic.2020-4-6 [ Links ]
62. Jamilloux Y, Henry T, Belot A, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev 2020;19(7):102567. https://doi.org/10.1016/j.autrev.2020.102567 [ Links ]
63. Molina JM, Delaugerre C, Le Goff J, et al. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe Covid-19 infection. Med Mal Infect 2020;50(384):30085-8. https://doi.org/10.1016/j.medmal2020.03.006 [ Links ]
64. Cortegiani A, Ingoglia G, Ippolito M, et al. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J Crit Care 2020;57(6):279-283. https://doi.org/10.1016/j.jcrc.2020.03.005 [ Links ]
65. Wessels I, Rolles B, Rink L. The potential impact of zinc supplementation on COVID-19 pathogenesis. Front Immunol 2020;11:1712. https://doi.org/10.3389/fimmu.2020.01712 [ Links ]
66. Marik PE, Kory P, Varon J. Does vitamin D status impact mortality from SARS-CoV-2 infection? Med Drug Discov 2020; 29 April(epub ahead of print). https://doi.org/10.1016/j.medidd.2020.100041 [ Links ]
67. Cheng RZ. Can early and high intravenous dose of vitamin C prevent and treat coronavirus disease 2019 (COVID-19)? Med Drug Discov 2020;5:100028. https://doi.org/10.1016/j.medidd.2020.100028 [ Links ]
68. Gupta A, Kashte S, Gupta M, Rodriguez HC, Gautam SS, Kadam S. Mesenchymal stem cells and exosome therapy for COVID-19: Current status and future perspective. Hum Cell 2020;33:907-918. https://doi.org/10.1007/s13577-020-00407-w [ Links ]
69. Galeotti C, Kaveri SV, Bayry J. IVIG-mediated effector functions in autoimmune and inflammatory diseases. Int Immunol 2017;29(11):491-498. https://doi.org/10.1093/intimm/dxx039 [ Links ]
70. Richards GA, Feldman C. The use of corticosteroids for COVID-19 infection. Afr J Thorac Crit Care Med. 2020;26(3):87-89. https://doi.org/10.7196/AJTCCM.2020.v26i3.10678 [ Links ]
71. Wu C, Chen X, Cai Y, et al Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med 2020;180(7):934-943. https://doi.org/10.1001/jamainternmed.2020.0994 [ Links ]
72. Montelongo-Jauregui D, Vila T, Sultan AS, Jabra-Rizk MA. Convalescent serum therapy for COVID-19: A 19th century remedy for a 21st century disease. PLoS Pathog 2020;16(8):e1008735. https://doi.org/10.1371/journal.ppat.100873 [ Links ]
73. Tang N, Bai H, Chen X, et al. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 2020;18(5):1094-1099. https://doi.org/10.1111/jth.14817 [ Links ]
74. Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): A review. JAMA 2020;323(18):1824-1836. https://doi.org/10.1001/jama.2020.6019 [ Links ]
75. Liu X, Cao W, Li T. High-dose intravenous immunoglobulins in the treatment of severe acute viral pneumonia: The known mechanisms and clinical effects. Front Immunol 14 July 2020. https://doi.org/10.3389/fimmu.2020.01660 [ Links ]
76. Horby P, Lim WS, Emberson JR, et al. Dexamethasone in hospitalized patients with Covid-19: Preliminary report. N Engl J Med 2020 Jul(epub ahead of print). https://doi.org/10.1056/NEJMoa2021436 [ Links ]
77. Alhazzani W, M0ller MH, Arabi YM, et al. Surviving Sepsis Campaign: Guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Intensive Care Med 2020;46(5):854-887. https://doi.org/10.1007/s00134-020-06022-5 [ Links ]
78. Feldstein LR, Rose EB, Horwitz SM, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med 2020;383(4):334-346 (epub ahead of print). https://doi.org/10.1056/NEJMoa2021680 [ Links ]
 Correspondence:
Correspondence:
M S Pepper
michael.pepper@up.ac.za
Accepted 14 October 2020


{kind=link}