










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.110 n.3 Pretoria Mar. 2020
http://dx.doi.org/10.7196/samj.2020.v110i3.14200
RESEARCH
A retrospective description of primary immuno-deficiency diseases at Red Cross War Memorial Children's Hospital, Cape Town, South Africa, 1975 - 2017
S MoodleyI; E GoddardII, III; M LevinIV, V; C ScottVI; A van EyssenVII; A DavidsonVIII, IX; R De DeckerX, XI; J M WilmshurstXII, XIII; A SpitaelsXIV, XV; B EleyXVI, XVII
IMB ChB, DCH (SA), Dip HIV Man (SA), FC Paed (SA); Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
IIMB ChB, PhD, MMed (Paed), FC Paed (SA), Cert Gastroenterology (SA) Paed; Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
IIIMB ChB, PhD, MMed (Paed), FC Paed (SA), Cert Gastroenterology (SA) Paed; Paediatric Gastroenterology Unit, Red Cross War Memorial Children's Hospital, Cape Town, South Africa
IVMB ChB, FC Paed (SA), MMed (Paed), Dip Allergy (SA), PhD; Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
VMB ChB, FC Paed (SA), MMed (Paed), Dip Allergy (SA), PhD; Division of Allergology, Red Cross War Memorial Children's Hospital, Cape Town, South Africa
VIMB ChB, FC Paed (SA), Grad Cert Paed Rheum (UWA); Paediatric Rheumatology Unit, Red Cross War Memorial Children's Hospital, Cape Town, South Africa
VIIMB ChB, DCH (SA), FC Paed (SA), CMO Paed (SA); Haematology/Oncology Service, Red Cross War Memorial Children's Hospital, Cape Town, South Africa
VIIIMB ChB, DCH (SA), FC Paed (SA), CMO Paed (SA), MPhil; Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
IXMB ChB, DCH (SA), FC Paed (SA), CMO Paed (SA), MPhil; Haematology/Oncology Service, Red Cross War Memorial Children's Hospital, Cape Town, South Africa
XMSc, MB ChB, DCH (UK), FC Paed (SA), Cert Med Genet (SA); Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
XIMSc, MB ChB, DCH (UK), FC Paed (SA), Cert Med Genet (SA); Paediatric Cardiology Unit, Red Cross War Memorial Children's Hospital, Cape Town, South Africa
XIIMB BS, MRCP, FC Paed (SA), MD; Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
XIIIMB BS, MRCP, FC Paed (SA), MD; Paediatric Neurology Unit, Red Cross War Memorial Children's Hospital, Cape Town, South Africa; and Neuroscience Institute, University of Cape Town, South Africa
XIVMB ChB, DCH, FC Paed (SA); Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
XVMB ChB, DCH, FC Paed (SA); Paediatric Endocrinology Unit, Red Cross War Memorial Children's Hospital and Groote Schuur Hospital, Cape Town, South Africa
XVIMB ChB, FC Paed (SA), BSc Hons; Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
XVIIMB ChB, FC Paed (SA), BSc Hons; Paediatric Infectious Diseases Unit, Red Cross War Memorial Children's Hospital, Cape Town, South Africa
ABSTRACT
BACKGROUND. The primary immunodeficiency diseases (PIDs) constitute a diverse and ever-expanding group of inborn errors affecting a wide range of immune functions. They are not well documented in sub-Saharan Africa.
OBJECTIVES. To describe the spectrum of PIDs at a tertiary paediatric hospital.
METHODS. A retrospective descriptive study of PIDs diagnosed at Red Cross War Memorial Children's Hospital, Cape Town, South Africa (SA), between 1975 and 2017 was undertaken.
RESULTS. We identified 252 children with PIDs, spanning eight of the nine categories listed in the 2017 classification of the International Union of Immunological Societies. Predominantly antibody deficiencies, combined immunodeficiencies with associated syndromic features, and immunodeficiencies affecting cellular and humoral immunity accounted for most children with PIDs (n= 199, 79.0%). The mean age (standard deviation) at diagnosis was 46 (50) months, and the male/female ratio was 1.5:1. There was a history of parental consanguinity in 3 cases (1.2%). Recurrent infection was the most prevalent presenting phenotype, manifesting in 177 patients (70.2%). Genetic or chromosomal confirmation was obtained in 42/252 cases (16.7%). Common interventions used to prevent infection were antimicrobial prophylaxis and immunoglobulin replacement therapy, administered to 95 (37.7%) and 93 (36.9%) of the patients, respectively. Six of 7 children who underwent haematopoietic stem cell transplantation (HSCT) had successful outcomes. The 7th patient died 2 months after HSCT from overwhelming infection. Although we could not account for the children lost to follow-up during the study period, 53 deaths were confirmed (21.0%).
CONCLUSIONS. Several challenges exist in the recognition and treatment of children with PIDs in our setting. These include limited access to genetic diagnostics and HSCT. Suboptimal treatment options contribute to the overall mortality of PIDs in SA.
Primary immunodeficiency diseases (PIDs) are inborn errors of immunity, a large heterogeneous group of predominantly genetic conditions that predispose individuals to a wide spectrum of infection, autoimmunity, autoinflammation, lymphoproliferation, malignancy, clinically recognisable syndromes in which immunodeficiency is a feature, allergic manifestations, and end-organ damage causing chronic dysfunction. Without optimal treatment, many PIDs are life-limiting.[1] Diagnosis is dependent on the type of infection and other presenting manifestations preceding diagnosis, immunological testing, and confirmatory genetic testing where possible.[2]
The spectrum of PIDs is evolving rapidly, with new PIDs continually being described and molecularly characterised. Since 1970, the International Union of Immunological Societies (IUIS) has included 354 PIDs and 344 different gene defects in its consensus classification.[3] Worldwide, PIDs affect more than six million people, and in Africa an estimated 988 000 adults and children have PIDs, of whom approximately 2 500 have been diagnosed.[4] In South Africa (SA), the estimated total population in mid-2017 was 56.52 million.[5] If it is assumed that the prevalence of PIDs in SA is similar to that in well-resourced countries, the total number of adults and children with PIDs in our country should range between 3 040 and 48 775. However, fewer than 500 PID cases have been reported from SA.[6,7]
Children with PIDs have been managed at Red Cross War Memorial Children's Hospital (RCWMCH) in Cape Town for more than 40 years.[8] An early review of the immunology laboratory database at the hospital during a 13-year period identified 93 children
with PIDs, a mean of 6.6 new PID diagnoses per annum. Primary antibody deficiencies predominated, accounting for 56% of all PIDs, and the spectrum of PIDs was similar to that reported from Europe and North and South America.[9] An analysis of 168 children with PIDs managed over a 27-year period at RCWMCH showed that the mean age at the time of diagnosis declined significantly over the study period, suggesting that awareness of these diseases among clinicians had improved.[6]In contrast to the situation in North Africa, where PIDs frequently occur in children with consanguineous parents, consanguinity was very unusual in the RCWMCH cohort, occurring in less than 2% of the patients.[6,10-12] High background consanguinity rates in North Africa and the Middle East alter the spectrum of PIDs compared with other geographical regions, with autosomal recessive conditions being more frequent than X-linked or autosomal dominant diseases.[13]
The current IUIS classification of PIDs includes a broad spectrum of conditions.[3] At RCWMCH several subspecialist services therefore participate in the management of children with PIDs, including rheumatology, haematology and oncology, cardiology, allergology, pulmonology, gastroenterology and neurology. Previous descriptive studies from RCWMCH have been confined to PID patients managed by the immunology and infectious diseases services.[9,6]
Objectives
To provide a more comprehensive understanding of the spectrum of PIDs across all subspecialist services at our hospital and raise awareness of PIDs among SA clinicians.
Methods
Study design and setting
This retrospective, descriptive study was completed at RCWMCH, a 273-bed, tertiary referral facility in Western Cape Province, affiliated to the University of Cape Town. It accepts paediatric referrals from various sources in both primary and tertiary settings, providing subspecialist consultation to doctors throughout SA but mainly from facilities managing children in the Western Cape. A dedicated service for the diagnosis and treatment of PIDs was established at RCWMCH in the mid-1970s.[14] In 2008 the PID service became part of the infectious diseases service at the hospital. Since 1983, information on all children with PIDs (i) who were diagnosed and/or treated, or (ii) for whom diagnostic/treatment advice was provided to their attending clinicians by the PID or infectious diseases services was maintained in a dedicated database, henceforth referred to as the PID database. Because of the multisystem nature of many PIDs, some children are increasingly being managed by subspecialist services other than the infectious diseases service at RCWMCH and were therefore not included in the PID database. To address this gap in the PID database and describe the full spectrum of PIDs managed or supported by all subspecialist services at RCWMCH between 1 January 1975 and 31 December 2017, the infectious diseases service collaborated with several specialist/ subspecialist services managing children with PIDs.
Classification of PIDs
Patients were classified according to the IUIS 2017 report on inborn errors of immunity and the IUIS 2017 phenotypic classification,[3,15] adapted where necessary using Stiehm's textbook on immune deficiencies.[16] Because mutational analysis is not routinely available in SA, most PID diagnoses are based on the clinical and immunological phenotype, and the nomenclature used to describe specific PIDs was therefore appropriately adapted.[6] Furthermore, diagnostic criteria formulated by the Pan-American Group for Immunodeficiency and the European Society for Immunodeficiency were used to diagnose some conditions such as immunoglobulin A deficiency and common variable immunodeficiency[17]
Data collection
Relevant clinical and laboratory data were extracted from hospital folders and the National Health Laboratory Service (NHLS) database using a standardised case record form that is used by the infectious diseases service in the management of patients with PIDs. Information included patient demographics, presenting problems, results of immunological investigation, treatment modalities, complications, and the outcome if known. The data were transferred anonymously in a study-specific Excel spreadsheet, version 2016 (Microsoft, USA).
Data analysis
Data were analysed using Stata statistical software, release 13 (StataCorp, USA). Proportions were expressed as percentages. Continuous variables were tested for normality using the Shapiro-Wilks test. Means and standard deviations (SDs) or medians and interquartile ranges were used to describe the data, as appropriate.
Ethics considerations
The research protocol was approved by the Human Research Ethics Committee, Faculty of Health Sciences, University of Cape Town (ref. no. 191/2017) and the RCWMCH research committee. Patient consent was not obtained, because the analysis was done retrospectively. The study was completed in accordance with the Declaration of Helsinki.
Results
Patient population
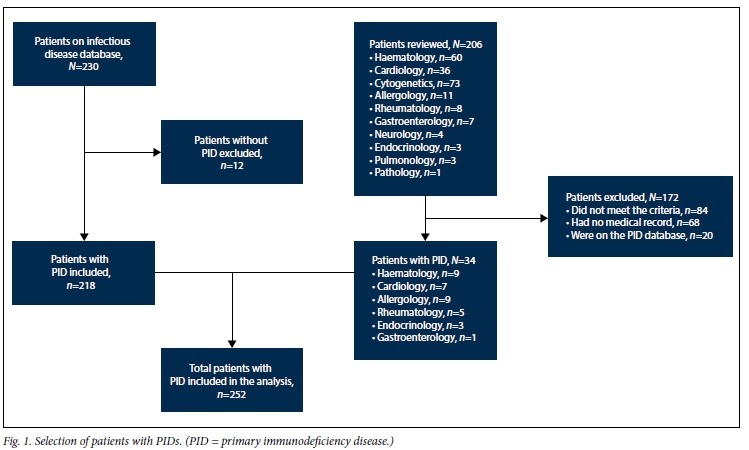
All children entered into the existing PID database during the study period were reviewed to confirm the accuracy of their diagnoses. Of 230 children in the database reviewed, 12 were excluded because there was no conclusive evidence that they had PIDs. The remaining 218 with PIDs were retained in the study database. Ten other specialist/subspecialist services referred 206 children with possible PIDs. These 206 patients were derived from outpatient attendance lists, clinical databases, the NHLS cytogenetics database and senior clinician memory. Clinical and laboratory records of these patients were reviewed, and a further 34 appropriately investigated children with PIDs were identified and added to the study database. A total of 252 children with PIDs were therefore identified and included in the current analysis (Fig. 1).
Spectrum of PIDs
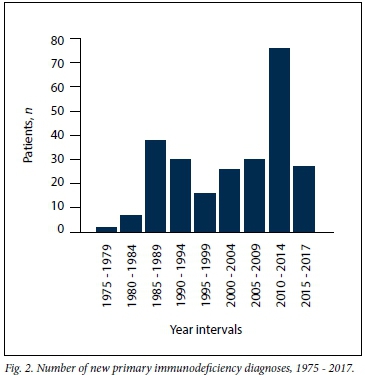
The number of new PID diagnoses in any 5-year period fluctuated throughout the study period, with no clear trend. The 2010 - 2014 period accounted for the highest number of new PID diagnoses (Fig. 2).
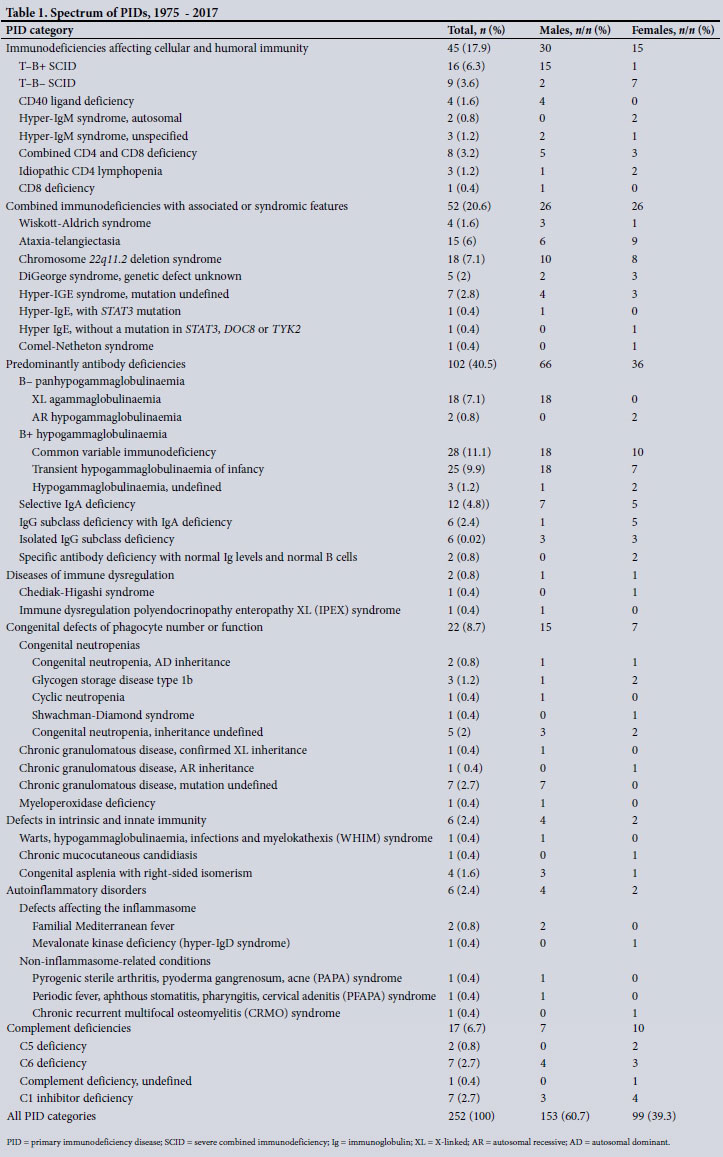
The spectrum of PIDs among the 252 children (Table 1) spanned eight of the nine categories listed in the 2017 IUIS report on inborn errors of immunity. Among the 252 patients, predominantly antibody deficiencies, combined immunodeficiencies with associated syndromic features, and immunodeficiencies affecting cellular and humoral immunity were most prevalent (Table 1), accounting for 79.0% of PIDs included in the study.
Patient characteristics
The mean (SD) age at the time of diagnosis was 46 (50) months. The male/female ratio was 1.5:1. There were more males than females in five of the eight PID categories (Table 1). Thirty-six patients (14.3%) were known to have a family history of PIDs. A history of parental consanguinity was present in 3 of the children (1.2%), one each with chronic granulomatous disease, severe combined immunodeficiency (SCID) caused by a mutation in recombination-activating gene 1 (RAG1) deficiency, and glycogen storage disease type lb. The majority of the patients (n=181, 71.8%), lived in the Western Cape, and a substantial number of patients were in the public health sector (Table 2).

Clinical presentation
Recurrent infection was the commonest presenting phenotype, manifesting in 177 patients (70.2%). Atypical infection and failure to thrive manifested in 38 (15.8%) and 51 (20.2%) patients, respectively. Invasive meningococcal infection was the presenting feature in 9 children with complement component deficiencies, of whom 6 manifested with recurrent meningococcal infection and 3 with a single episode of infection before diagnosis. One child was diagnosed after screening based on a family history of complement C5 deficiency. Dysmorphic features or recognisable clinical syndromes occurred in 52 patients (20.6%). The three commonest syndromes were DiGeorge syndrome, formerly called 22q11.2 deletion syndrome, ataxia-telangiectasia and hyper-IgE syndrome, occurring in 23 (9.1%), 15 (6.2%) and 9 (3.6%) of the patients, respectively (Table 1). Six of the 7 children with CI inhibitor deficiency presented with angio-oedema, while the remaining patient, an older brother of one of the other patients, was screened based on the family history and diagnosed before he manifested clinically. The 6 children with autoinflammatory disorders presented with non-infectious manifestations, commonly persistent fever.
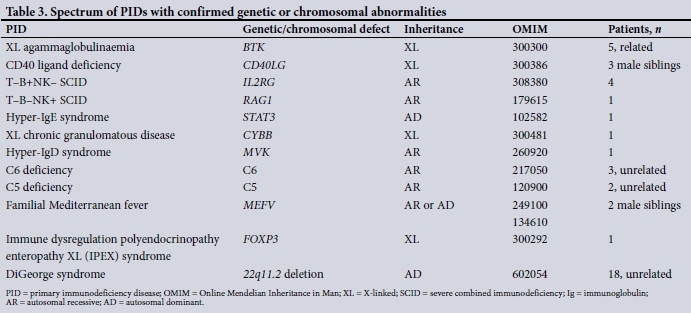
Genetic or chromosomal confirmation
Molecular characterisation is not routinely available in SA, but was undertaken in a subset of the patient cohort. Single-gene mutations were confirmed in 24 patients in the cohort (9.5%) and chromosomal deletions in a further 18 (7.1%) (Table 3).
Management
The predominant interventions administered to prevent infection were antimicrobial prophylaxis (95/252, 37.7%) and immunoglobulin replacement therapy (93/252, 36.9%). Antimicrobial prophylaxis plus immunoglobulin replacement therapy was administered to 31/252 patients (12.3%). Seven children underwent haematopoietic stem cell transplantation (HSCT). The first HCST, for a child with SCID caused by interleukin-2 receptor gamma-chain deficiency, took place in 1996 (Brian Eley, personal communication 23 July 2019). Six of the 7 patients had SCID and one immune dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome. Six of these patients experienced complete immunological reconstitution and are well. The 7th patient, who underwent HSCT for underlying SCID, died 2 months after HSCT from presumed septicaemia complicated by multiorgan failure. Other treatment interventions included subcutaneous interferon-gamma administration in 2 patients with chronic granulomatous disease. The treatment of patients with CI inhibitor deficiency included fresh-frozen plasma during acute attacks and tranexamic acid prophylaxis. In the patients with autoinflammatory disorders, treatment included colchicine (n=3), methotrexate (n=2) and adalimumab, an anti-tumour necrosis factor-alpha inhibitor (n=1).
Mortality
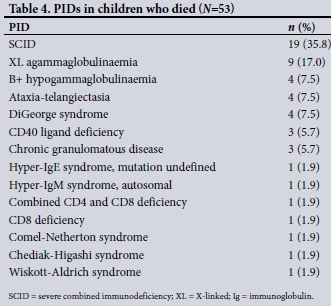
During the study period, 53 patients (21.0%) died. The cause of death was not known in 12 cases. In the remaining 41 patients, the most frequent causes of death were presumed or proven septicaemia (n=15, 37.5%) and pneumonia (n=9, 22.5%). Among 47 patients with a known date of death, death occurred within 1 year of PID diagnosis in 32 cases (68.1%), including 15 (31.9%) who died within 1 month of diagnosis. Of the remainder, 7 (14.9%) died between 1 and 5 years after PID diagnosis and 8 (17.0%) more than 5 years after diagnosis (Table 4).
Discussion
Worldwide, PIDs are an ever-expanding disease group as a result of the description of new genetic disorders. The burden of secondary immunodeficiencies such as HIV infection and severe acute malnutrition continues to overshadow that of the PIDs in low- to middle-income countries (LMICs), including SA, resulting in limited investment in the diagnostic and treatment resources needed for PIDs. Despite the advancing knowledge base and expertise on PIDs, national incidence and prevalence estimates are largely unknown. An important barrier to care is limited awareness of PIDs and their management among healthcare professionals, as well as in the broader society. A study completed in the USA and published in 2016 showed that compared with subspecialist immunologists, few family practitioners were aware of guidelines for diagnosing and managing PIDs (4% v. 79%).[18] In the present study, we documented the spectrum of PIDs among 252 children diagnosed at a tertiary hospital in SA over a period of more than 40 years. Although diagnosis depended mainly on clinical and immunological criteria, we were able to classify our patients using the latest IUIS classification. The spectrum of PIDs in this study spanned eight of the nine categories in the IUIS classification. The ninth category, phenocopies of inborn errors of immunity, was only added to the IUIS classification very recently, in 2015, and many of the diseases in this category manifest in adulthood, hence the absence of these PIDs among our patients.[3]
Parental consanguinity was identified in only 1.2% of our cases.[13] In contrast, in the North African countries of Morocco, Tunisia and Egypt, parental consanguinity rates of 43.2%, 58.2% and 62.5%, respectively, were documented.[10-12] These high parental consanguinity rates result in higher proportions of PIDs due to Mendelian autosomal recessive inheritance, lower proportions of PIDs due to Mendelian X-linked inheritance, and consequently a predominance of combined cellular and humoral immunodeficiency compared with the preponderance of predominantly antibody deficiencies in populations with low consanguinity rates.[13] An analysis of the European Society for Immunodeficiencies (ESID) registry in June 2014 showed that 56.8% of 19 355 children and adults with PIDs had predominantly antibody deficiencies.[19] In our study, predominantly antibody deficiencies accounted for 40.5% of all PIDs, whereas in the registries of Morocco and Tunisia, predominantly antibody immunodeficiencies accounted for 22.8% and 17.7% of all PIDs, respectively.[11,12]
The mean age of our patients was 46 months, similar to the mean age of children in the Egyptian study of 51 months.[10] The mean ages at diagnosis in the Moroccan and Tunisian registries were 6.2 years and 5.3 years, respectively[11,12] However, both these registries included children and adults with PIDs. A previous analysis from our hospital published in 2011 documented a mean age at diagnosis of 51 months and a statistically significant decline in mean age over time, suggesting that awareness of the PIDs had increased among clinicians at our hospital during the study period.[6] Although most of our patients emanated from the public sector, 36.9% were from the private sector.
Given that approximately 20% of the SA population are serviced in the private sector, this result suggests that more effort may be needed to increase awareness of the PIDs among public sector clinicians.
Genetic or chromosomal confirmation of PID diagnosis was obtained in 16.7% of our patients, somewhat higher than 13.8% genetic confirmation reported in 2015 in the Tunisian registry[11] In Europe, where molecular testing is more accessible, the rate of genetic diagnostic confirmation is much higher, having reached 36% of all patients registered with ESID registry in April 2018.[20]
Most interventions for managing PIDs are available to a varying degree in SA, including optimal immunisation practice, antimicrobial therapy and prophylaxis, immunoglobulin replacement therapy and infection control measures.[2] Some interventions, such as interferon-1 inhibitors and HSCT, are extremely expensive, limiting access in the public sector. HSCT is the treatment of choice for many severe PIDs. Our findings demonstrate that it is possible to undertake HSCT successfully in patients with PIDs in SA. However, only a small proportion of more than 50 patients who could have benefited received a transplant. Reasons for the low number of transplants include late presentation of patients, often with severe complications of their underlying PID, the small donor pool in SA, resulting in the unavailability of suitable matched unrelated donors, high cost
of accessing donors affiliated to international registries, and no paediatric intensive care facilities at the adult tertiary referral hospital where our paediatric transplants were undertaken. In 2012, the first HLA-haploidentical HSCT was performed for SCID. Before 2012, only HLA-identical or matched unrelated donors were considered for children with SCID. Transplantation programmes have also been started in North African countries such as Tunisia and Morocco.[11,12] Unfortunately we had limited access to patient outcomes, especially for those lost to follow-up. We are, however, able to describe the mortality of 21.0%, which is comparable to mortality documented in studies with similar challenges completed in Egypt, Morocco and Tunisia of 23.4%, 28.8% and 34.5%, respectively[10-12]In our study;36% of known deaths occurred in patients with SCID, and 68% of deaths, including 19 preceded by SCID, occurred during the first year after PID diagnosis, indicating the need to focus on optimising the outcome of children with SCID because there is a high chance of achieving immunological cure after successful HSCT. In the Tunisian study, 17.2% of patients were lost to follow-up, suggesting that this may be an important outcome in patients with PIDs in other LMICs and should be quantified.[11]
Study strengths and limitations
This study is the largest single-centre description of PIDs in children in sub-Saharan Africa to date. We were able to include more than 250 patients because we adopted an expanded search strategy among subspecialist services at our hospital. Furthermore, despite limited genetic confirmation, we were able to classify our patients using the latest IUIS classification system. Because of the retrospective study design, there were limitations in the availability and completeness of the clinical data. Outcome data were limited to known deaths. Because some of the patients were treated by their referring doctors with minimal or no feedback, the true mortality prevalence was not established and we therefore did not explore risk factors associated with death. We could not comprehensively document those who were lost to follow-up, and so have an incomplete understanding of the true extent of this outcome measure among our patients. Review of patients referred by other subspecialist services showed that a sizeable proportion were not adequately investigated for immunodeficiency. For example, of 73 patients with laboratory-confirmed 22q11 deletion, 37 (50.7%) had not been investigated for immunodeficiency and therefore could not be considered for inclusion in our study population. Likewise, of 15 patients with right-sided isomerism, 11 had not been investigated for asplenia. Furthermore, it is likely that more cases of PID were missed during the study period because either PID was not considered or the children presented with milder PID phenotypes. Although this reality prevented us from achieving complete description of PIDs at our hospital during the study period, our results did allow us to largely address the stated objective and provide direction for improving our clinical service.
Conclusions
In the present study, we were able to collate the findings of 252 patients with a spectrum of 50 primary immunodeficiency diseases over a period of more than four decades. Greater awareness among clinicians treating children and more laboratory diagnostic capacity are needed to increase the recognition of PIDs among children in SA. The treatment options that are available in SA are unevenly distributed. Treatment capacity should therefore be expanded throughout the country, especially advanced interventions such as HSCT Ongoing reporting of registries like ours and increased community awareness should strengthen the lobby for greater investment in rare diseases such as the PIDs.
Declaration. The research for this study was done in partial fulfilment of the requirements for SM's MMed degree at the University of Cape Town.
Acknowledgements. Spasina King, Simoné Twaku and Michelle Kannemayer are acknowledged for retrieval of patient medical records and data extraction. Adelaide Masu assisted with the data analysis.
Author contributions. SM designed the study, wrote the protocol, analysed the data and wrote the initial draft of the article. BE conceived the study and assisted with the study design, protocol development, data analysis and drafting of the article. All authors contributed patients to the study provided critical comment on the draft article and approved the final submitted article.
Funding. None.
Conflicts of interest. None.
References
1. Modell V, Orange JS, Quinn J, et al Global report on primary immunodeficiencies. 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol Res 2018;66(3):367-380. https://doi.org/10.1007/s12026-018-8996-5 [ Links ]
2. Eley B, Esser M. Investigation and management of primary immunodeficiency in South African children. S Afr Med J 2014;104(11):793. https://doi.org/10.7196/SAMJ.8946 [ Links ]
3. Picard C, Gaspar BH, Al-Herz, W, et al. International Union of Immunological Societies. 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity J Clin Immunol 2018;38:96-128. https://doi.org/10.1007/s10875-017-0464-9 [ Links ]
4. Bousfiha AA, Jeddane L, Erwa N, et al. Development of primary immunodeficiencies in Africa. J Clin Immunol 2015;35(4):329-330. https://doi.org/10.1007/s10875-015-0162-4 [ Links ]
5. Statistics South Africa. Mid-year population estimates 2017. Statistical release P0302. Pretoria. Stats SA, 2017. http://www.statssa.govza/publications/P0302/P03022017.pdf (accessed 7 December 2017). [ Links ]
6. Naidoo R Ungerer L, Cooper M, Pienaar S, Eley B. Primary immunodeficiencies. A 27-year review at a tertiary paediatric hospital in Cape Town, South Africa. J Clin Immunol 2011;31:99-105. https://doi.org/10.1007/s10875-010-9465-7 [ Links ]
7. Esser M. Primary immunodeficiency. Missed opportunities and treatment challenges. Curr Allergy Clin Immunol 2012;25(4):184-188. https://journals.co.za/content/caci/25/4/EJC130086 (accessed 5 February 2020). [ Links ]
8. Haddad HL, Beatty DW, Dowdle EB. Chronic granulomatous disease of childhood. S Afr Med J 1976;50:2068-2072. [ Links ]
9. Eley BS, Hughes J, Cooper M, et al. Primary immunodeficiency diseases at Red Cross War Memorial Children's Hospital. S Afr Med J 1997;87(12):1684-1688. [ Links ]
10. Reda SM, Ami HM, Amine MM. Primary immunodeficiency diseases in Egyptian children. A single-center study J Clin Immunol 2009;29(3):343-351. https://doi.org/10.1007/s10875-008-9260-x [ Links ]
11. Mellouli F, Mustapha IB, Khaled MB, et al. Report of the Tunisian registry of primary immunodeficiencies. 25-years of experience (1988 - 2012). J Clin Immunol 2015;35(8):745-753. https://doi.org/10.1007/s10875-015-0206-9 [ Links ]
12. Bousfiha AA, Jeddane L, El Hafidi N, et al. First report on the Moroccan registry of primary immunodeficiencies. 15 years of experience (1998 - 2012). J Clin Immunol 2014;34(4):459-468. https://doi.org/10.1007/s10875-014-0005-8 [ Links ]
13. Barbouche MR, Mekki N, Ben-Ali M, Ben-Mustapha I. Lessons from genetic studies of primary immunodeficiencies in a highly consanguineous population. Front Immunol 2017;8.737. https://doi.org/10.3389/fimmu.2017.00737 [ Links ]
14. Eley B, Beatty D. Primary immunodeficiency diseases in Cape Town. Allergy Clin Immunol Int 2000;12(6):267-270. [ Links ]
15. Bousfiha A, Jeddane L, Picard C, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol 2018;38:129-143. https://doi.org/10.1007/s10875-017-0465-8 [ Links ]
16. Sullivan KE, Stiehm R, eds. Stiehnis Immune Deficiencies. Elsevier, 2015;1113. https://doi.org/10.1016/B978-0-12-405546-9.00054-6 [ Links ]
17. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol 1999;93(3):190-197. https://doi.org/10.1016/C2012-1-01317-3 [ Links ]
18. Orange JS, Seeborg FO, Boyle M, et al. Family physician perspectives on primary immunodeficiency diseases. Front Med 2016;3:12. https://doi.org/10.3389/fmed.2016.00012 [ Links ]
19. Mahlaoui N, Gathmann B, Kindle G, et al. The European Society for Immunodeficiencies (ESID) Registry. Recent advancements in the epidemiology of primary immunodeficiencies and how does that translate into clinical practice. Rare Dis Orphan Drugs 2014;1(4 Suppl 4):25-27. https://esid.org/content/download/14136/395600/file/EPIRARE_RareJournal%20Dec%202014_ESID_REGISTRY.pdf (accessed 5 February 2020). [ Links ]
20. Mahlaoui N. European Society for Immunodeficiency registry working party newsletter, April 2018. https://esid.org/Working-Parties/Registry-Working-Party/ESID-Registry/Registry-WP-Newsletter-April-2018 (accessed 5 December 2018). [ Links ]
 Correspondence:
Correspondence:
S Moodley
sashmi.moodley@yahoo.com
Accepted 29 July 2019


{kind=link}
{kind=link}
{kind=link}