










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.110 no.2 Pretoria feb. 2020
http://dx.doi.org/10.7196/samj.2020.v110i2.14594
CME
N A AlliI; J VaughanII; S LouwII
IMB BCh, FCPath (SA) Haem;Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg; and National Health Laboratory Service, Johannesburg, South Africa
IIMB BCh, MMed (Haem), FCPath (SA) Haem; Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg; and National Health Laboratory Service, Johannesburg, South Africa
IIIMB BCh, MMed (Haem), FCPath (SA) Haem, PhD; Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg; and National Health Laboratory Service, Johannesburg, South Africa
ABSTRACT
Thromboembolic conditions are the leading cause of mortality, estimated to account for 1 in 4 deaths worldwide in 2010. Over time, the incidence and mortality rates of these conditions have improved in developed countries, but are increasing in developing countries. The haemostatic system comprises 6 main components, i.e. (i) platelets; (ii) vascular endothelium; (iii) coagulation proteins; (iv) natural anticoagulants; (v) the fibrinolytic system; and (vi) natural antifibrinolytic factors. A delicate balance exists between procoagulant and anticoagulant factors within the vascular system. Numerous acquired or inherited conditions may tip the balance either way, i.e. towards a prothrombotic or prohaemorrhagic state. In this issue of CME, the first of a 2-part series on thrombophilic disorders, the subject of discussion is on inherited varieties that the general practitioner is likely to encounter. This review is primarily based on venous thrombosis.
Haemostasis is a physiological process that stops blood loss at the site of injury, while maintaining normal blood flow in the rest of the circulation.
This is accomplished in three physiological steps that occur in rapid sequence: (i) vasoconstriction; (ii) formation of a platelet plug (primary haemostasis); and (iii) stabilisation of the clot through cross-linking of insoluble fibrin (secondary haemostasis). Apart from limiting blood loss, the clot allows for vessel and tissue repair.
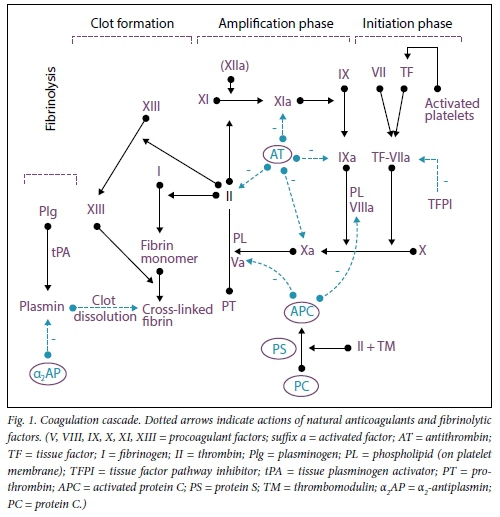
Anticoagulant mechanisms regulate the coagulation system to ensure formation of a clot that is proportional to the injury. A delicate balance between procoagulant and anticoagulant systems is critical for proper haemostasis and for avoiding pathological bleeding or thrombosis. The clot is finally dissolved by the fibrinolytic system, which also performs the function of preventing blood clots in healthy blood vessels (Fig. 1). Any disturbance to the vessel wall, vascular flow or balance between procoagulant and anticoagulant molecules in the blood (i.e. Virchow's triad) may result in thrombosis.[1] Thrombotic disorders may be categorised as either arterial or venous, where the symptoms and pathophysiological processes are quite distinct. Arterial thrombosis is usually preceded by damage to the arterial wall. Symptoms include pain at the site of hypoxia and/or loss of function distal to the site of obstruction due to ischaemia/ infarction. Venous thrombosis may cause discomfort or pain and is often accompanied by oedema distal to the thrombus. Large-vein thromboses (calf and popliteal veins) carry a higher risk of pulmonary embolism.
The focus of this article (part 1 of a 2-part CME series) is inherited thrombophilia, where diagnostic and pathophysiological aspects are discussed. Acquired thrombo-philia is discussed in part 2 (next issue). Management and prevention of thrombosis are beyond the scope of this CME; we refer the reader to the local guidelines for more information in this regard.[2]
Inherited thrombophilia due to abnormalities of natural anticoagulants
Antithrombin deficiency
Antithrombin (AT), a serine protease, is a key regulator of the coagulation system. It is a natural inhibitor of active clotting factors, particularly thrombin and factor X (FX) and, to a lesser extent, FXII, FXI, FIX and FVII. Its complete absence from the body is incompatible with life, and the natural inhibitory activity of AT is accelerated by the conformational change that occurs when bound to heparin.[3] AT further regulates inflammation in the vascular endothelium, controls microvascular leakage and inhibits pro-inflammatory cytokines. Inadequate activity levels of AT in plasma increase the risk of thrombotic events, both in the venous and, to a lesser extent, arterial circulation, and deficiencies can be inherited or acquired. AT is produced mainly in the liver, but also in endothelial cells. Congenital AT deficiency constitutes the most significant inherited thrombophilic condition, with an odds ratio of up to 20 for the development of venous thrombosis in the heterozygous state, which is increased further in the presence of acquired risk factors, such as pregnancy and malignancy. More than 250 mutations in the AT gene have been reported. Population-wide screening of this condition, albeit controversial, occurs in an estimated 0.2% of the population.[4] Occurrence of a first thrombotic event in patients with congenital deficiency is related more to the type of genetic mutation, the presence of concomitant acquired predisposing conditions and the patient's age than to AT plasma levels.
Acquired AT deficiency occurs in a variety of physiological and pathological conditions with similar risks of increased thrombosis.[5] The mechanism of AT depletion in acquired deficiencies relates to reduced hepatic synthesis, increased consumption, abnormal leakage from the circulation, blood dilution and the effect of drugs.[6] Acquired causes of AT deficiency include pregnancy, ovarian hyperstimulation syndrome, liver failure, nephrotic syndrome, inflammatory bowel disease, the postsurgical period, sepsis, thermal injury, heparin therapy and massive venous thrombosis. Long-term anticoagulation therapy is often indicated after a thrombotic event, and in high-risk situations AT concentrates are a treatment option for patients with congenital AT deficiency.[4]
Protein C and protein S deficiency
Protein C (PC) is a vitamin K-dependent natural anticoagulant produced mainly in the liver. PC is activated by thrombin via its interaction with thrombomodulin and endothelial PC receptor in the presence of protein S (PS) (Fig. 1). Activated PC inactivates membrane-bound activated coagulation FV and FVIII.[7] PC has a plasma half-life of 6 - 8 hours. Congenital PC deficiency is caused by >160 mutations thus far identified in the PC gene.[8] Heterozygous PC deficiency occurs in ~0.5% of the general population and in 3 -9% of random patients with a first venous thromboembolic event (VTE). The risk of thrombosis in congenital PC deficiency increases further in association with other risk factors, such as pregnancy.[9,10] Homozygous PC deficiency results in severely low PC levels and can cause intrauterine fetal death or present with purpura fulminans and disseminated intravascular coagulation in the newborn, which may necessitate long-term anticoagulation and replacement therapy with fresh frozen plasma or PC concen-rates.[11,12] Congenital PC deficiency can be either quantitative (type I) or qualitative (type II), and laboratory assays are available for distinguishing between these subtypes. Acquired PC deficiency is more common than inherited genetic abnormalities and relates to vitamin K deficiency, which occurs with oral anticoagulant therapy and liver synthetic dysfunction. Acquired causes must be excluded prior to considering a congenital cause of low PC levels.[11,12]
PS is also a vitamin K-dependent factor produced in the liver and endothelial cells, which enhances the anticoagulant activity of PC, and its deficiency is associated with an increased thrombotic risk. PS in the plasma has a half-life of 42 hours and exists in two forms, i.e. PS bound to complement 4b-binding protein (60%), which is the inactive PS component, and free PS (40%), which is the physiologically active component. These two components comprise the total PS in the plasma. Congenital PS deficiency is estimated to be present in <0.5% of the general white population and 2 - 12% of selected groups with venous thromboembolic disease, and consists of type I (reduced total and free antigen), type II (normal total and free antigen, but reduced activity) and type III (normal total antigen, but reduced free antigen resulting in reduced activity) deficiencies. Type I and type III account for 95% of cases of congenital PS deficiency.[7,8] Acquired causes of PS deficiency are encountered more frequently and relate to vitamin K deficiency due to treatment with vitamin K antagonists (e.g. warfarin), liver disease, severe and chronic inflammation, autoimmune syndromes, nephrotic syndrome, pregnancy and viral infections, such as HIV.
Patients with congenital PC or PS deficiency have a 2 - 11-fold increased risk of a VTE (deep-vein thrombosis (DVT) with or without pulmonary embolism (PE)) in comparison with unaffected individuals, although the final occurrence of a VTE depends on multiple underlying genetic and acquired factors. Patients with PC/PS deficiency may manifest with cerebral, visceral or axillary vein thrombosis, but these thromboses are less common. PC/PS-deficient patients are also at risk of warfarin-induced skin necrosis.[8] Some studies have suggested an association between PC/PS deficiencies and arterial thromboses, such as ischaemic stroke, but this is not conclusive.[12] The development of thrombosis in PC/ PS-deficient patients may occur in association with other congenital conditions, such as the FV Leiden mutation, or provoked by acquired conditions such as pregnancy.[7] The estimated annual incidenceof a VTE in PC-deficient (0.7%) and PS-deficient (0.8%) patients is higher than in those with the FV Leiden mutation. Provoked venous thromboembolic episodes in patients with PC/PS deficiencies are labelled as low-risk events and are treated with short-term anticoagulation for 3 - 6 months. Heparin should be administered for at least 1 day prior to the introduction of warfarin to prevent skin necrosis. Patients with idiopathic unprovoked events are considered at high risk for VTE recurrence and may benefit from lifelong anticoagulation. Ongoing thromboprophylaxis is not recommended in asymptomatic individuals with PC/PS deficiencies.[13]
Activated protein C resistance
A single nucleotide substitution in the FV gene, known as the FV Leiden mutation, is a common genetic abnormality that predisposes carriers to a VTE.[14] This mutation renders FV relatively resistant to cleavage by activated PC, thereby promoting thrombin generation. Additional rare point mutations have been described, i.e. FV Hong Kong and FV Cambridge, which are also associated with activated PC (APC) resistance and increased risk of venous thromboembolism. The risk of a VTE is increased 2 - 7-fold in heterozygotes and 40 - 80-fold in individuals homozygous for FV Leiden.[15] FV Leiden is common in the white population, with a prevalence of 3 - 7%. However, the mutation has not been associated with thrombosis among black population affinity groups.[16]
FV Leiden testing is performed by functional testing for APC resistance or by DNA testing for the Arg506Gln mutation. The optimal management of carriers and their relatives should be guided by familial penetrance, characteristics of the first VTE (e.g. provoked/unprovoked, age) and laboratory biomarkers.[17]
Inherited thrombophilia due to increased thrombotic potential
Prothrombin mutation
The prothrombin polymorphism G20210A is a common genetic abnormality that predisposes carriers to a VTE. The polymorphism is characterised by increased prothrombin activity, which is the precursor of thrombin. Similar to the FV Leiden mutation, the prothrombin gene mutation is also prevalent in white populations (0.7 - 4.0%).[18] Several studies have shown that this polymorphism is a low-to-moderate risk factor for a VTE.[19] The risk of a VTE is increased 2 - 3-fold in heterozygotes and up to 19-fold in homo-zygotes. The polymorphism is an important risk factor in conjunction with the FV Leiden mutation or acquired thrombotic risk factors (e.g. oral contraceptive therapy, pregnancy, immobilisation, surgery).[20]
Prothrombin gene testing is performed by DNA testing for G20210A. The optimal management of carriers and their relatives should be guided by familial penetrance, characteristics of the first VTE (e.g. provoked/unprovoked, age) and the presence of acquired or additional inherited thrombotic risk factors.[20]
Increased coagulation factor levels
Coagulation factors play a central role in the initiation and amplification of secondary clot formation. Increased coagulation factor levels, i.e. FII, FVII, FVIII, FIX, FX, FXI, fibrinogen and Von Willebrand factor, are associated with an increased VTE risk.[13,21] In contrast, elevated levels of FV, FXII and FXIII have not been associated with a VTE. FVIII has been shown to be the strongest risk factor.[21] In the coagulation cascade, activated FVIII, which is the cofactor of FIX, potentiates the generation of thrombin. The molecular mechanism underlying elevated FVIII levels is, however, not clear.[22] It is likely that elevated FVIII levels alone are insufficient to cause a VTE, but that the risk is increased in the presence of other causal factors.
Testing for FVIII is often included as part of a thrombotic screen in patients with venous thromboembolic disease. As FVIII is an acute-phase reactant, it is important to perform testing after the thrombotic acute-phase response. Increased FVIII levels have been detected in ~25% of patients with VTEs. Owing to the increased risk of recurrent VTEs, high FVIII levels can be used to risk stratify patients for extended thromboprophylaxis.[23]
Increased plasminogen activator inhibitor-1 levels
The primary mediator of fibrinolysis is tissue plasminogen activator (tPA). Plasminogen activator inhibitor-1 (PAI-1) is the primary inhibitor of tPA. It is released from endothelial cells, platelets, monocytes and hepatocytes in response to inflammatory stimuli or upregulation of thrombin. Family studies have reported a 4G/5G polymorphism, located in the promoter region of the PAI-1 gene, to be associated with elevated PAI-1 levels.[24] Increased levels of PAI-1 with impaired fibrinolysis predispose to a VTE. However, studies of the role of elevated levels of PAI-1 in patients with venous thromboemblic disease have shown conflicting results.[25] This suggests that elevated PAI-1 is insufficient to cause a VTE, but that the risk is marginally increased in the presence of other causal factors, including inflammation or other prothrombotic conditions.[26]
Further studies are indicated before treatment recommendations can be made. Genetic testing for the 4G/5G polymorphism and PAI-1 levels may be used for diagnostic purposes, but are not routinely available.
Dysfibrinogenaemia
Dysfibrinogenaemia encompasses a group of disorders that may be inherited or acquired and is characterised by normal or reduced fibrinogen antigen levels, but disproportionately low fibrinogen activity levels (qualitative fibrinogen defect).[27] Acquired conditions are most commonly encountered in the context of liver dysfunction. The inherited varieties are rare. Patients may present with bleeding, a VTE (often at a young age) or poor obstetric history. However, most individuals are asymptomatic and are usually identified by the prolongation of screening coagulation tests. It has been proposed that abnormal fibrinogen predisposes to a VTE, either as a result of defective binding with thrombin, and/or decreased tPA-mediated fibrinolysis by plasmin. Dysfibrinogenaemia is characterised by prolongation of the prothrombin time, activated partial thromboplastin time (APTT) and thrombin time. The diagnosis of dysfibrinogenaemia is based on the assessment of functional and antigenic fibrinogen. Specialised testing, such as genetic analysis, is also required.[28]
Recommendations for patient management are derived from expert consensus guidelines.[29] Management should be considered in conjunction with a personal and family history of bleeding or thrombosis. Specific management in the setting of surgery and pregnancy requires a multidisciplinary team. Close follow-up of asymptomatic patients is advised owing to the risk of complications during the natural course of the disease.
Miscellaneous inherited thrombophilias
Protein Z deficiency
Protein Z (PZ) is a vitamin K-dependent factor with homology to other vitamin K-dependent factors such as prothrombin, PC and PS, but it lacks enzymatic activity. Instead, PZ acts as a cofactor for the PZ-dependent protease inhibitor (ZPI), which inhibits FXa. PZ deficiency is postulated to be associated with a procoagulant state, playing a pathogenic role in arterial and venous thromboembolic diseases.[30] An association between altered plasma PZ concentrations and adverse pregnancy outcomes, including recurrent miscarriage and pre-eclampsia, has been demonstrated. The mechanism of PZ deficiency in pregnant patients, with resultant obstetric complications, may relate to anti-PZ autoantibodies.[31] The roles of PZ plasma activity and PZ gene polymorphisms, however, remain poorly defined, with conflicting results of studies, where some report low PZ levels in association with an increased risk of venous thrombosis and pregnancy complications, whereas others did not observe this association.[32] Routine measurement of PZ levels does not currently form part of the laboratory assessment of thrombosis risk.
Tissue factor pathway inhibitor
Tissue factor pathway inhibitor (TFPI), produced in endothelial cells, regulates the tissue factor (TF) pathway. The process of coagulation commences with TF, which binds and activates FVII. This results in the production of thrombin (FIIa) on the surface of a TF-bearing cell. This initial generation of thrombin is very short lived, as TF:FVIIa is very rapidly inactivated by TFPI. Lower TFPI levels are associated with increased thrombin generation. Low plasma levels of TFPI have been observed in patients with venous thromboembolic disease and stroke.[33-35] However, a significant association between low TFPI levels and an increased risk of a VTE has not been consistently demonstrated.
Testing for TFPI is not routinely available. To date, there is no strong evidence to suggest that TFPI deficiency should be considered a clinically significant prothrombotic condition. Further studies are indicated before treatment recommendations can be made.
Thrombomodulin deficiency
Thrombomodulin is a surface receptor expressed by intact healthy endothelial cells and binds thrombin that has diffused away from the site of endothelial damage. In addition to blocking the procoagulant function of thrombin, the binding of thrombin to thrombomodulin brings it into close proximity of PC, leading to inactivation of FV and FVIII. Consequently, mutations of the thrombomodulin gene (THBD) have been considered a risk factor for a VTE. These mutations are, however, rare and evidence from clinical trials suggests that these are not significant risk factors for venous thromboembolic disease.[36,37] These polymorphisms are predominantly seen in African-American populations, in contrast to FV Leiden and prothrombin G20210A gene mutations.[38]
Testing for mutations of the thrombomodulin gene is not routinely available. There is to date no strong evidence to suggest that these mutations should be considered a clinically significant prothrombotic condition. Further studies are indicated before treatment recommendations can be made.
Hyperhomocysteinaemia
Homocysteine is an intermediate product of methionine metabolism. Levels of homocysteine are regulated by the enzyme, methylenetetrahydrofolate reductase (MTHFR). Genetic mutations of this enzyme result in hyperhomocysteinaemia and an increased risk of cardiovascular disease. In addition, homocysteine levels are influenced by ethnic and regional differences, reduced folic acid, vitamin B12 and vitamin B6 levels, lifestyle, medical conditions and drugs. Homocysteine levels >15 μηιοΙ/L are defined as hyperhomocysteinaemia. Mild hyperhomocysteinaemia is a common laboratory finding. While the role of homocysteine in arterial disease has been well researched, its role in venous thromboembolic disease is controversial. It is not clear from the available evidence whether hyperhomocysteinaemia is the cause or consequence of a VTE. A meta-analysis concluded that hyperhomocysteinaemia is a weak risk factor for a VTE.[39]
Currently, there is insufficient evidence to support the testing of fasting homocysteine levels in patients with venous thromboembolic disease.[40] Furthermore, there is insufficient evidence for vitamin supplementation in hyperhomocysteinaemia, in particular with folic acid and vitamin B12.[41] Vitamin supplementation, however, is advised in patients with folate or vitamin B12 deficiency. Patients should be educated regarding the uncertain role of this treatment in the prevention of recurrence of a VTE.
Laboratory testing
FV Leiden, prothrombin gene mutations and antiphospholipid antibodies, which will be discussed in part 2 of this CME series, constitute the most prevalent thrombophilias. Deficiencies of the natural anticoagulants (PC, PS and AT) and elevated homocysteine levels have a lower yield and should generally be limited to patients with high-risk and unique presentations. Patients with unprovoked splanchnic vein thrombosis should also be assessed for myeloproliferative neoplasms and paroxysmal nocturnal haemoglobinuria. Selection criteria for appropriate VTE testing are listed in Table 1.

It should be noted that first-degree relatives of patients with a prior VTE are at elevated risk, even if thrombophilia testing results are negative in the index patient. Thrombophilia testing need not be done in patients with a clear provoking event, such as recent major orthopaedic surgery in the absence of other factors suspicious for thrombophilia or in cases of underlying malignancy, as results are unlikely to alter the treatment plan.[42]
Timing of sample collection
Samples for FV Leiden and prothrombin gene mutation, as well as serological antiphospholipid antibodies such as anticardiolipin antibodies, can be collected in the acute phase of thrombosis, as the results are not affected by acute thrombosis or anticoagulants. Functional lupus anticoagulant testing in the coagulation laboratory, and testing for PC, PS and AT deficiencies, should be delayed until completion of anticoagulation therapy. However, in patients who might require lifelong anticoagulation, samples should be collected in the acute phase prior to initiation of therapy.
Conclusions
Venous thromboembolic disease is an important cause of morbidity and mortality. In southern Africa, the HIV epidemic has unexpectedly caused a concurrent VTE epidemic, which has not been recognised. Clinicians in southern Africa need to become more cognisant of this devastating disease. It is hoped that this review helps to address the problem. Patients who have had a VTE can be assessed and the majority of inherited and acquired disorders can be diagnosed. This will assist the clinician in deciding on the duration of anticoagulation, as well as help determine whether family studies are indicated to prevent a possible devastating thrombosis.
Declaration. None.
Acknowledgements. None.
Author contributions. NAA: concept, design and content; JV: design and content; SL, ES, BJ: content.
Funding. None.
Conflicts of interest. None.
References
1. Bagot CN, Arya R. Virchow and his triad: A question of attribution. Br J Haematol 2008;143(2):180-190. https://doi.org/10.1111/j.1365-2141.2008.07323.x [ Links ]
2. Jacobson BF, Louw S, Buller H, et al. Venous thromboembolism: Prophylactic and therapeutic guideline. S Aft Med J 2013;103(4):260-267. https://doi.oig/10.7196/SAMJ.6706 [ Links ]
3. Muszbek L, Bereczky Z, Kovacs B, Komaromi I. Antithrombin deficiency and its laboratory diagnosis. Clin Chem Lab Med 2010;48(Suppl 1):S67-S78. https://doi.org/10.1515/CCLM.2010.368 [ Links ]
4. Bauer KA, Nguyen-Cao TM, Spears JB. Issues in the diagnosis and management ofhereditary antithrombin deficiency. Ann Pharmacother 2016;50(9):758-767. https://doi.org/10.1177/1060028016651276 [ Links ]
5. Ornaghi S, Barnhart KT, Frieling J, Streisand J, Paidas MJ. Clinical syndromes associated with acquired antithrombin deficiency via microvascular leakage and the related risk of thrombosis. Thromb Res 2014;133(6):972-984. https://doi.org/10.1016/j.thromres.2014.02.014 [ Links ]
6. Pal N, Kertai MD, Lakshminarasimhachar A, Avidan MS. Pharmacology and clinical applications of human recombinant antithrombin. Expert Opin Biol Ther 2010;10(7):1155-1168. https://doi.org/10.1517/14712598.2010.495713 [ Links ]
7. Wypasek E, Undas A. Protein C and protein S deficiency - practical diagnostic issues. Adv Clin Exp Med 2013;22(4):459-467. [ Links ]
8. Ten Kate MK, van der Meer J. Protein S deficiency: A clinical perspective. Haemophilia 2008;14(6):1222-1228. https://doi.org/10.111/j.1365-2516.2008.01775.x [ Links ]
9. Miletich J, Sherman L, Broze G, Jr. Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med 1987;317(16):991-996. https://doi.org/10.1056/NEJM198710153171604 [ Links ]
10. Vossen CY, Conard J, Fontcuberta J, et al. Familial thrombophilia and lifetime risk of venous thrombosis. J Thromb Haemost 2004;2(9):1526-1532. https://doi.org/10.1111/j.1538-7836.2004.00852.x [ Links ]
11. Khor B, van Cott EM. Laboratory tests for protein C deficiency. Am J Hematol 2010;85(6):440-442. https://doi.org/10.1002/ajh.21679 [ Links ]
12. Mahmoodi BK, Brouwer JL, Veeger NJ, van der Meer J. Hereditary deficiency of protein C or protein S confers increased risk of arterial thromboembolic events at a young age: Results from a large family cohort study. Circulation 2008;118(16):1659-1667. https://doi.org/10.1161/CIRCULATIONAHA.108.780759 [ Links ]
13. Goldhaber SZ, Piazza G. Optimal duration of anticoagulation after venous thromboembolism. Circulation 2011;123(6):664-667. https://doi.org/10.1161/CIRCULATIONAHA.110.970822 [ Links ]
14. Dahlback B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: Prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA 1993;90:1004-1008. https://doi.org/10.1073/pnas.90.3.1004 [ Links ]
15. Rosendaal F, Koster T, Vandenbroucke J, et al. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995;85(6):1504-1508. [ Links ]
16. Ridker PM, Miletich JP, Hennekens CH, et al. Ethnic distribution of factor V Leiden in 4 047 men and women. Implications for venous thromboembolism screening. JAMA 1997;277(16):1305-1307. [ Links ]
17. Campello E, Spiezia L, Simioni P. Diagnosis and management of factor V Leiden. Expert Rev Hematol 2016;9(12):1139-1149. https://doi.org/10.1080/17474086.2016.1249364 [ Links ]
18. Rosendaal FR, Reitsma PH. Genetics of venous thrombosis. J Thromb Haemost 2009;7(Suppl 1):301-304. https://doi.org/10.1111/j.1538-7836.2009.03394.x [ Links ]
19. Bank I, Libourel EJ, Middeldorp S, et al. Prothrombin 20210A mutation: A mild risk factor for venous thromboembolism but not for arterial thrombotic disease and pregnancy-related complications in a family study. Arch Intern Med 2004;164(17):1932-1937. https://doi.org/10.1001/archinte.164.17.1932 [ Links ]
20. Simone B, de Stefano V, Leoncini E, et al. Risk of venous thromboembolism associated with single and combined effects of factor V Leiden, prothrombin 20210A and methylenetetrahydrofolate reductase C677T: A meta-analysis involving over 11 000 cases and 21 000 controls. Eur J Epidemiol 2013;28(8):621-647. https://doi.org/10.1007/s10654-013-9825-8 [ Links ]
21. Rietveld IM, Lijfering WM, le Cessie S, et al. High levels of coagulation factors and venous thrombosis risk: Strongest association for factor VIII and Von Willebrand factor. J Thromb Haemost 2019;17(1):99-109. https://doi.org/10.1111/jth.14343 [ Links ]
22. Jenkins PV, Rawley O, Smith OP, et al. Elevated factor VIII levels and risk of venous thrombosis. Br J Haematol 2012;157(6):653-663. https://doi.org/10.1111/j.1365-2141.2012.09134.x [ Links ]
23. Timp JF, Lijfering WM, Flinterman LE, et al. Predictive value of factor VIII levels for recurrent venous thrombosis: Results from the MEGA follow-up study. J Thromb Haemost 2015;13(10):1823-1832. https://doi.org/10.1111/jth.13113 [ Links ]
24. Vuckovic BA, Djeric MJ, Tomic BV, et al. Influence of decreased fibrinolytic activity and plasminogen activator inhibitor-1 4G/5G polymorphism on the risk of venous thrombosis. Blood Coag Fibrinol 2018;29(1):19-24. https://doi.org/10.1097/MBC.0000000000000656 [ Links ]
25. Sundquist K, Wang X, Svensson PJ, et al. Plasminogen activator inhibitor-1 4G/5G polymorphism, factor V Leiden, prothrombin mutations and the risk of VTE recurrence. Thromb Haemost 2015;114(6):1156-1164. https://doi.org/10.1160/TH15-01-0031 [ Links ]
26. Tsantes AE, Nikolopoulos GK, Bagos PG, et al. The effect of the plasminogen activator inhibitor-1 4G/5G polymorphism on the thrombotic risk. Thromb Res 2008;122(6):736-742. https://doi.org/10.1016/j.thromres.2007.09.005 [ Links ]
27. De Moerloose P, Casini A, Neerman-Arbez M. Congenital fibrinogen disorders: An update. Semin Thromb Hemost 2013;39(6):585-595. https://doi.org/10.1055/s-0033-1349222 [ Links ]
28. Shapiro SE, Phillips E, Manning RA, et al. Clinical phenotype, laboratory features and genotype of 35 patients with heritable dysfibrinogenaemia Br J Haematol 2013;160(2):220-227. https://doi.org/10.1111/bjh.12085 [ Links ]
29. Mumford AD, Ackroyd S, Alikhan R, et al. Guideline for the diagnosis and management of the rare coagulation disorders: A United Kingdom haemophilia centre doctors' organization guideline on behalf of the British Committee for Standards in Haematology. Br J Haematol 2014;167(3):304-326.https://doi.org/10.1111/bjh.13058 [ Links ]
30. Broze GJ, Jr. Protein-Z and thrombosis. Lancet 2001;357(9260):900-901. https://doi.org/10.1016/S0140-6736(00)04229-X [ Links ]
31. Almawi WY, Al-Shaikh FS, Melemedjian OK, Almawi AW. Protein Z, an anticoagulant protein with expanding role in reproductive biology. Reproduction 2013;146(2):R73-R80. https://doi.org/10.1530/REP-13-0072 [ Links ]
32. Bafunno V, Santacroce R, Margaglione M. The risk of occurrence of venous thrombosis: Focus on protein Z. Thromb Res 2011;128(6):508-515. https://doi.org/10.1016/j.thromres.2011.08.007 [ Links ]
33. Fei X, Wang H, Yuan W, et al. Tissue factor pathway inhibitor-1 is a valuable marker for the prediction of deep venous thrombosis and tumor metastasis in patients with lung cancer. Biomed Res Int 2017;2017:8983763. https://doi.org/10.1155/2017/8983763 [ Links ]
34. Dahm A, van Hylckama Vlieg A, Bendz B, et al. Low levels of tissue factor pathway inhibitor (TFPI) increase the risk of venous thrombosis. Blood 2003;101(11):4387-4392. https://doi.org/10.1182/blood-2002-10-3188 [ Links ]
35. Zakai NA, Lutsey PL, Folsom AR, et al. Total tissue factor pathway inhibitor and venous thrombosis. The longitudinal investigation of thromboembolism etiology. Thromb Haemost 2010;104(2):207-212. https://doi.org/10.1160/TH09-10-0693 [ Links ]
36. Ahmad A, Sundquist K, Zöller B, et al. Thrombomodulin gene c.1418C>T polymorphism and risk of recurrent venous thromboembolism. J Thromb Thrombolysis 2016;42(1):135-141. https://doi.org/10.1007/s11239-015-1328-x [ Links ]
37. Heit JA, Petterson TM, Owen WG, et al. Thrombomodulin gene polymorphisms or haplotypes as potential risk factors for venous thromboembolism: A population-based case-control study. J Thromb Haemost 2005;3(4):710-717. https://doi.org/10.1111/j.1538-7836.2005.01187.x [ Links ]
38. Hernandez W, Gamazon ER, Smithberger E, et al. Novel genetic predictors of venous thromboembolism risk in African Americans. Blood 2016;127(15):1923-1929. https://doi.org/10.1182/blood-2015-09-668525 [ Links ]
39. Ray JG, Shmorgun D, Chan WS. Common C677T polymorphism of the methylenetetrahydrofolate reductase gene and the risk of venous thromboembolism: Meta-analysis of 31 studies. Pathophysiol Haemost Thromb 2002;32(2):51-58. https://doi.org/10.1159/000065076 [ Links ]
40. Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic therapy and prevention of thrombosis. Evidence-based clinical practice guidelines. Chest 2012; 141(2 Suppl):e419S-e494S. https://doi.org/10.1378/chest.11-2301 [ Links ]
41. Bonaa KH, Njolstad I, Ueland PM, et al. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med 2006;354(15):1578-1588. https://doi.org/10.1056/NEJMoa055227 [ Links ]
42. Carroll BJ, Piazza G. Hypercoagulable states in arterial and venous thrombosis: When, how, and who to test? Vasc Med 2018;23(4):388-399. https://doi.org/10.1177/1358863X18755927 [ Links ]
 Correspondence:
Correspondence:
N A Alli
nazeer.alli@nhls.ac.za
Accepted 8 January 2020

