










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.109 n.11 Pretoria Nov. 2019
http://dx.doi.org/10.7196/samj.2019.v109i11.13889
RESEARCH
Elevated unidentified antibodies in sickle cell anaemia patients receiving blood transfusions in Cape Town, South Africa
K A ThompsonI; F AdamsII; G M DavisonIII, IV
IBHSc; Western Province Blood Transfusion Service, Pinelands, Cape Town, South Africa
IIMSc; Western Province Blood Transfusion Service, Pinelands, Cape Town, South Africa
IIIPhD (Haematology); Department of Biomedical Sciences, Faculty of Health and Wellness Sciences, Cape Peninsula University of Technology, Cape Town, South Africa
IVPhD (Haematology); Division of Haematology, Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
BACKGROUND. Sickle cell disease (SCD) is an inherited haemoglobinopathy in which homozygous individuals suffer from numerous complications including vaso-occlusion, infection and haemolytic anaemia. Patients therefore often require blood transfusions, which may lead to elevated levels of allogeneic antibodies. In South Africa (SA), the number of patients with SCD has risen significantly owing to migration and changing demographics, leading to an increased need for blood products.
OBJECTIVES. Against the above background, to determine the incidence of alloimmunisation and the presence of clinically significant antibodies in SCD patients requiring transfusions in Cape Town, SA.
METHODS. Information on SCD patients receiving blood products between 2010 and 2012, including demographics, number of units transfused and the presence and type of alloantibodies, was collected. The results were compared with those for non-SCD controls who had received a similar number of transfusions.
RESULTS. Of 182 patients analysed, 91 had SCD. Twenty-two percent of the SCD patients and 13.2% of the controls had become alloimmunised (p=0.12), while the proportions of those receiving transfusions and acquiring clinically significant antibodies were similar between the two groups (p=0.17 and p=0.19, respectively). However, the total number and amount of unidentified antibodies were significantly increased in patients with SCD (p=0.02 and p<0.001, respectively).
CONCLUSIONS. This study concluded that patients with SCD develop increased numbers of unidentified antibodies, which may be important in the selection of suitable donors.
Sickle cell disease (SCD) is the most common inherited haemoglobin disorder.[1] This haemoglobinopathy is caused by a nucleotide base substitution (adenine - thymine) leading to an amino acid change from glutamic acid to valine on the beta-globin chain.[2] The amino acid substitution results in the formation of haemoglobin S (HbS), which has the potential of significantly reducing the lifespan of the red cell. In low oxygen tension environments HbS polymerises, causing elongation of the red cell, which takes on a 'sickle' shape. The normal red cell shape may be restored once an optimal oxygen environment returns, but after repeated episodes, the erythrocyte membrane becomes damaged and the sickle shape becomes irreversible.[3] The pathophysiology of SCD is complex, with patients suffering from a number of complications including vaso-occlusion and hypoxia leading to painful crises, increased infections, splenic dysfunction, haemolytic anaemia, aplastic crises and organ damage.[4]
While treatments such as hydroxyurea, gene therapy and stem cell transplantation have all been used with success, blood transfusion remains one of the most important forms of therapy.[5,6] The transfusion of blood products aids in restoring the number of normal red cells, improving oxygen-carrying capacity and blood flow, particularly during incidents of severe haemolysis.[7]
Although blood transfusions are an effective treatment for SCD, there are many risks including alloimmunisation, iron overload and transmission of blood-borne pathogens. The development of alloantibodies in the recipient, probably due to blood group antigenic differences between the donor and the recipient, can lead to delayed transfusion reactions that can be fatal.[8] Patients with SCD have been identified as having increased incidences of alloimmunisation despite full compatibility testing between donor and recipient. The most likely cause of this is the receipt of blood products from donors whose red cell antigens are phenotypically different to those of SCD patients, who are mainly of black African descent.[5]
Objectives
The incidence of SCD in South Africa (SA) has significantly increased as a result of migration and changing demographics. Consequently, the need for blood transfusions and blood products for this group of patients has escalated.[9] Cape Town has a diverse population, and as genetic disparity between donor and recipient is the most likely cause of alloantibody development, a study examining the rate of alloimmunisation in patients with SCD receiving blood products in Cape Town was considered important.
Our objectives were to analyse the frequency and distribution of red cell antibodies occurring in SCD patients who had received red blood cell products in Western Cape Province and to compare them with those in a group of non-SCD patients who had received a similar number of transfusions.
Methods
Study design and patient selection
This was a retrospective, cross-sectional study of 182 patients and controls who had received a minimum of one blood transfusion. Data from individuals receiving transfusions between 2010 and 2012 were collected from the Western Province Blood Transfusion Service (WPBTS). Information regarding age, clinical diagnosis, number of transfusions and antibodies was collected from the WPBTS's Phoenix database and stored on an Excel spreadsheet, version 2010 (Microsoft, USA). Patients who had required adult red blood cell concentrate, infant red blood cell concentrate, paediatric red blood cell concentrate, leucocyte-reduced red blood cell product or whole blood unit/s were included, while individuals both with and without SCD who had suffered from a recent viral or bacterial infection were excluded. The data from SCD patients were compared with those from a group of randomly selected non-SCD patients who had received a similar number of transfusions.
Ethical approval
Ethical approval was obtained from the Faculty of Health and Wellness Sciences Ethics Committee at the Cape Peninsula University of Technology (ref. no. CPUT/HW-REC 2016D1). Patient information was kept confidential and was anonymised by allocating a number to each patient and removing any personal information such as name, hospital number and identification number. Data collected were not shared with any unauthorised personnel and were stored on a password-protected computer that was only available to the principal investigator.
Data collection
Historical records of patients with 'Sickle cell disease' or 'Sickle cell anaemia' or 'SCA' or 'SCD' or 'Hb SS' or 'Hb SC' were collected. Information including patient age, blood group, antibodies and number of units issued was recorded on an Excel spreadsheet, version 2010. Similar data were collected from a group of patients who had also received blood but did not have SCD. This control group was made up of randomly selected trauma, obstetric and leukaemia patients. As extended phenotyping was started in 2013, data were collected from 2010 to 2012 to avoid any bias and to reflect the true alloimmunisation rate of SCD patients before extended phenotyping began.
Blood grouping and antibody identification
ABO and Rh blood grouping as well as antibody screens were performed on the AutoVue or BioVue systems (Ortho Clinical Diagnostics, USA) using known cells or antisera supplied by Ortho Clinical Diagnostics. If antibody screens were positive, patient samples were sent to the WPBTS Reference Laboratory where the presence of any irregular antibodies was determined using a panel of 12 known-donor red blood cell suspensions to identify antibody or antibodies by a process of elimination. Each of the 12 donor cell suspensions was incubated with patient plasma, and if it was reactive with one of the cell suspensions expressing a particular antigen, the patient was identified as having developed an antibody to the corresponding antigen. If the reaction did not match the expected pattern of expression across the 12 donor cell suspensions, the antibody was referred to as unidentified. The presence of an antibody known to cause transfusion reactions and/or haemolytic disease of the fetus or newborn was considered clinically significant.
Statistical analysis
Data were recorded on an Excel spreadsheet, version 2010, and analysed using Statistica version 13 (Dell, USA). A p-value <0.05 was considered significant when comparing the experimental group against the control data. The parameters were analysed for significance using descriptive analysis (means, ranges and frequencies), Student's f-test and the χ2 test. Graphs and tables were created using Excel 2010.
Results
Patient demographics and blood groups
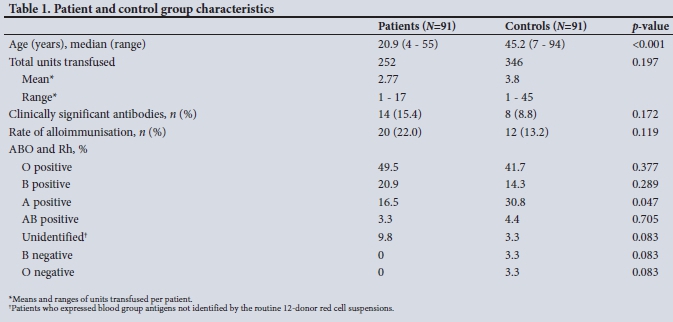
A total of 182 patients who had received at least one blood transfusion were studied. Of these, 91 (50%) had SCD. The other 91 (50%) were trauma, obstetric and leukaemia patients who had received at least one blood transfusion but did not have SCD. The mean age of the SCD patient group was 20.9 years (range 4 -55), which was significantly lower than that of the control group (mean 45.2 years, range 7 - 94). Although the SCD patients had an increased incidence of alloimmunisation and development of clinically significant antibodies, this was not statistically different from the control group. The distribution of ABO blood group and Rh antigens was similar across the two groups, but patients with SCD had a significantly lower incidence of blood group A Rh positivity (p=0.047), The demographic data and blood group results are shown in Table 1.
Frequency of alloantibodies
The alloimmunisation rate with clinically significant antibodies was higher in the SCD patients (15.4%) than in the control group (9.9%), but this difference was not significant (p=0.172). Regardless of antibody type, 22.0% of the patient group and 13.2% of the control group became alloimmunised (p=0.119), with the median number of transfusions being higher in the control group (3.8) than in the patient group (2.8). However, a significantly increased number of total antibodies was detected in the SCD patients compared with the controls (p=0.016), and the number of unidentified antibodies present in the SCD group v. the controls was also significantly elevated (p<0.001) (Table 2).
Discussion
The objectives of this study were to document the frequency and distribution of allogeneic red cell antibodies in SCD patients receiving blood products in the Western Cape. Although the rate of alloimmunisation was similar to that of the controls (p=0.119), the results revealed significantly elevated numbers of total antibodies (p=0.016), of which many were unidentified (p<0.001).
Of the 91 SCD patients, 22.0% had been alloimmunised, and although this figure was not significantly different to that of the control group, it was higher than reported in many other studies in the literature, which range from 6.9% to 74%.[10-13] The rate of alloimmunisation is influenced by a number of factors including history of pregnancy, number of blood transfusions, age at first transfusion, sex and genetics.[5,14-16] In a study of SCD patients who had been fully cross-matched,[17] it was reported that individuals receiving more than 8.9 units of blood had a significantly higher alloimmunisation rate (59%) than those receiving less (15%). In our study, the median number of transfused units in SCD patients (2.77) was not significantly different to that in the control group (3.8), but the median age of the SCD patients was significantly lower (p<0.001). Many younger children have underdeveloped immune responses, and this may influence the recognition of foreign antigens and the consequent development of allogeneic antibodies.[18] Since their underdeveloped immune systems may not recognise donor antigens, it is therefore envisaged that as these children age and receive more transfusions, the rate of alloimmunisation will increase.
Despite the lower number of transfused units, the patients with SCD had a significantly increased number of total allogeneic antibodies (p=0.016), of which many were unidentified (p<0.001). These data must be interpreted in context, however, as although there was an increase in unidentified antibodies, the total number of clinically significant alloantibodies in the two groups was not statistically different. Many of the detected antibodies were unidentified and had possibly developed to antigens not usually seen in our region.
SCD is prevalent throughout sub-Saharan Africa, the Middle East and India.[19] The majority of patients with SCD in Cape Town originate from Africa, in particular the Democratic Republic of the Congo.[9] One of the major causes of the development of allogeneic antibodies and alloimmunisation is the difference in the antigenic phenotype between the donor and the recipient.[5] In areas where there is ethnic diversity and the donor population is racially different to the recipients, the alloimmunisation rate is significantly increased.[20] This theory appears to be supported by other studies in countries with more homogeneous populations such as Uganda, where alloimmunisation rates are lower at 6%.[21]
According to the 2011 population census of the Western Cape,[22] the population had increased by 28% since the previous census in 2001 and the increase was largely attributed to migration from other provinces and outside the country. The Western Cape has a total of 5.8 million people, which can be broken down into those of mixed ancestry (48.8%), black Africans (32.8%), Asians (2%) and whites (15%). In contrast, although the WPBTS recruits donors across all ethnic groups residing in the Western Cape, the profile of donors is very different to the demographics of the province, and in particular those suffering from SCD. The WPBTS integrated report of 2017 -2018[23] indicated that 59% of donors were white, 34% of mixed ancestry, 1% Asian and 6% of black African descent. The increased number of unidentified antibodies may therefore be linked to antigenic differences resulting from the genetic and ethnic diversity in the region.
Patients with SCD have a number of additional factors that could predispose them to developing alloantibodies. These include clinical complications, an abnormal immune response, and polymorphisms of the immune regulatory gene TRIM21.[5,16]In addition, SCD has often been described as a chronic inflammatory disorder with increased inflammatory cytokines that could influence the rate of alloimmunisation and the development of autoantibodies.[24] In our study there was no difference in the development of autoantibodies between the two groups. Tests evaluating levels of inflammation were not available, so this association could not be made.
The result of alloimmunisation is the development of delayed haemolytic transfusion reactions due to the reaction of allogeneic antibodies to antigens expressed on the donor red cells. These antibodies are often not detected prior to the transfusion owing to low titres and lack of transfusion records. In addition, it is thought that the proinflammatory environment present in SCD promotes increased haemolysis of the donor cells and causes the patient's sickled red cells to adhere to the vessel wall, causing vaso-occlusion.[5] These reactions can be life threatening and may be missed as they often occur up to 2 weeks after the transfusion has taken place. It is therefore important for clinicians to monitor patients who have received blood products carefully, and for blood transfusion services to implement strategies that attempt to prevent these reactions from occurring. These should include the transfusion of leucodepleted red cells and extended antibody screening. Recruitment of donors of black African descent and from population groups representative of patients with SCD is also required.
Study limitations
A limitation of this study was that full demographic and diagnostic information regarding the haemoglobin variant was not available. It is well recognised that certain haemoglobin variants are associated with increased alloimmunisation rates. For example, individuals with the homozygous HbSS and HbSb0 thalassaemia variants have higher incidences of alloimmunisation than those with HbSC.[25] In Cape Town, HbC and thalassaemia have been identified in individuals of mixed ancestry and in those of Indian and Mediterranean descent.[9] In order to understand the above findings fully, further studies would therefore need to include full demographic and diagnostic criteria.
Conclusions
Despite the above limitation, this study has been able to conclude that patients with SCD develop significantly elevated numbers of allogeneic antibodies, of which many are unidentified. The disparity between donor and patient red cell phenotypes is the most likely cause. The Western Cape has many diverse population groups, and as migration from other parts of the African continent is likely to continue, the incidence of diseases such as SCD will continue to rise. The blood transfusion services throughout SA have an important role to play in the treatment and survival of these patients, and will therefore be required to address these challenges. New strategies to recruit donors from all population groups, including those from Central Africa, and more comprehensive antibody screening may be required.
Since completion of this study, extended phenotyping for SCD has been introduced and further research investigating the impact of these strategies is under way.
Declaration. This publication and study formed part of the requirement for KAT's BHSc (Medical Laboratory Science) degree.
Acknowledgements. The authors thank the WPBTS for data retrieval and Dr Corrie Uys for her assistance with the statistical analysis.
Author contributions. KAT: substantial contributions to conception and design, analysis of data and approval of manuscript to be published. FA: substantial contributions to conception and design, analysis of results, supervision of KAT, and final approval of the version to be published. GMD: substantial contributions to conception and design, drafting the article, supervision of student and final approval of the version to be published.
Funding. None.
Conflicts of interest. None.
References
1. Ashley-Koch A, Yang Q, Olney R. Sickle hemoglobin (Hb S) allele and sickle cell disease: A HuGE review. Am J Epidemiol 2000;151(9):839-845. https://doi.org/10.1093/oxfordjournals.aje.a010288 [ Links ]
2. Natarajan K, Townes TM, Kutlar A. Disorders of hemoglobin structure: Sickle cell anemia and related abnormalities. In: Lichtman MA, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT, eds. Williams Hematology. 8th ed. New York: McGraw-Hill, 2010:1104-1165. [ Links ]
3. Laudicina RJ. Hemoglobinopathies: Qualitative defect. In: McKenzie SD, Williams JL, eds. Clinical Laboratory Hematology. London: Pearson, 2012:211-230. [ Links ]
4. Ware RE, de Montalembert M, Tshilolo L, et al. Sickle cell disease. Lancet 2017;390(10091):311-323. https://doi.org/10.1016/S0140-6736(17)30193-9 [ Links ]
5. Yazdanbakhsh K, Ware R, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: Pathophysiology, risk factors, and transfusion management. Blood 2012;120(3):528-537. https://doi.org/10.1182/blood-2011-11-327361 [ Links ]
6. Alli NA, Patel M, Alli HD, et al. Recommendations for the management of sickle cell disease in South Africa. S Afr Med J 2014;104(11):743-751. https://doi.org/10.7196/SAMJ.8470 [ Links ]
7. Blaney KD, Howard PR. Basic and Applied Concepts of Blood Banking and Transfusion Practices. 3rd ed. St Louis: Mosby, 2013:329-344. [ Links ]
8. Natukunda B, Brand A, Schonewille H. Red blood cell alloimmunization from an African perspective. Curr Opin Hematol 2010;17(6):565-570. https://doi.org/10.1097/moh.0b013e32833ec54b [ Links ]
9. Wonkam A, Ponde C, Nicholson N, et al. The burden of sickle cell disease in Cape Town. S Afr Med J 2012;102(9):752-754. https://doi.org/10.7196/SAMJ.5886 [ Links ]
10. Rosse WF, Gallagher D, Kinney TR, et al. Transfusion and alloimmunization in sickle cell disease: The Cooperative Study of Sickle Cell Disease. Blood 1990;76(7):1431-1437. http://www.bloodjournal.org/content/76/7/1431.long?sso-checked=true (accessed 11 September 2019). [ Links ]
11. Aly R, El-sharnoby MR, Hagag AA. Frequency of red cell alloimmunization in patients with sickle cell anemia in an Egyptian referral hospital. Transfus Apher Sci 2012;43(3):253-257. https://doi.org/10.1016/j.transci.2012.07.014 [ Links ]
12. Meda E, Magesa PM, Marlow T, et al. Red blood cell alloimmunization in sickle cell disease patients in Tanzania. East Afr Med J 2014;11(2). http://www.ajol.info/index.php/eajph/article/view/108580 (accessed 11 September 2019). [ Links ]
13. Elenga N, Niel L. Alloimmunization in patients with sickle cell disease in French Guiana. J Blood Transfus 2015; Article ID 812934. https://doi.org/10.1155/2015/812934 [ Links ]
14. Zimring JC, Hendrickson JE. The role of inflammation in alloimmunization to antigens on transfused red blood cells. Transfus Med Immunohematol 2008;15(6):631-635. https://doi.org/10.1097/moh.0b013e328313695e [ Links ]
15. Pandey H, Das SS, Chaudhary R. Red cell alloimmunization in transfused patients: A silent epidemic revisited. Asian J Transfus Sci 2014;8(2):75-77. https://doi.org/10.4103/0973-6247.137433 [ Links ]
16. Tatari-Calderone Z, Gordish-Dressman H, Fasano R, et al. Protective effect of HLA-DQB1 alleles against alloimmunization in patients with sickle cell disease. Hum Immunol 2016;77(1):35-40. https://doi.org/10.1016/j.humimm.2015.10.010 [ Links ]
17. Chou S, Jackson T, Vege S, et al. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors. Blood 2013;122(6):1062-1071. https://doi.org/10.1182/blood-2013-03-490623 [ Links ]
18. Harmening D. Modern Blood Banking and Transfusion Practices. Philadelphia: FA Davis Company, 2012. [ Links ]
19. Williams TN, Weatherhall DJ. World distribution, population genetics, and health burden of the haemoglobinopathies. Cold Spring Harb Perspect Med 2012;2:a011692. https://doi.org/10.1101/cshperspect.a011692 [ Links ]
20. Bauer MP, Wiersum-Osselton J, Schipperus M. Clinical predictors of alloimmunization after red blood cell transfusion. Transfusion 2007;47(11):2066-2071. https://doi.org/10.1111/j.1537-2995.2007.01433.x [ Links ]
21. Natukunda B, Schonewille H, Ndugwa C, et al. Red blood cell alloimmunisation in sickle cell disease patients in Uganda. Transfusion 2010;50:20-25. https://doi.org/10.1111/j.1537-2995.2009.02435.x [ Links ]
22. Statistics South Africa. 2011. Report-03-01-70 - Census 2011: Provincial Profile: Western Cape. Pretoria: Stats SA, 2015. http://www.statssa.gov.za/?page_id=1854&PPN=Report-03-01-70 (accessed 28 February 2016). [ Links ]
23. Western Province Blood Transfusion Service. Integrated Report, 2017 - 2018, p. 38. https://www.wcbs.org.za/?q=about/annual-reports (accessed 10 December 2018). [ Links ]
24. Fasano RM, Booth GS, Miles M. Red blood cell alloimmunization is influenced by recipient inflammatory state at time of transfusion in patients with sickle cell disease. Br J Haematol 2015;168(2):291-300. https://doi.org/10.1111/bjh.13123 [ Links ]
25. Telen MJ, Afenyi-Annan A, Garret ME, et al. Alloimmunization in sickle cell disease: Changing antibody specificities and association with chronic pain and decreased survival. Transfusion 2015;55(602):1378-1387. https://doi.org/10.1111/trf.12940 [ Links ]
 Correspondence:
Correspondence:
G M Davison
davisong@cput.ac.za
Accepted 23 April 2019


{kind=link}
{kind=link}