










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.109 n.10 Pretoria Oct. 2019
http://dx.doi.org/10.7196/samj.2019.v109i10.14376
CME
An approach to the diagnosis and management of multiple myeloma
F FazelI; F BassaII
IMB BCh, FCP (SA), MMed (Int Med), Cert Clin Haematology (SA); Division of Clinical Haematology, Department of Internal Medicine, Faculty of Medicine and Health Sciences, Tygerberg Academic Hospital and Stellenbosch University, Cape Town, South Africa
IIMB ChB, FC Path (Haem), MMed (Haem); Division of Clinical Haematology, Department of Internal Medicine, Faculty of Medicine and Health Sciences, Tygerberg Academic Hospital and Stellenbosch University, Cape Town, South Africa
ABSTRACT
Multiple myeloma (MM) is a plasma cell dyscrasia that accounts for ~10% of haematological malignancies. It is a disease of the elderly, with a slight male predominance. Almost all cases of MM are preceded by an asymptomatic, premalignant phase known as monoclonal gammopathy of undetermined significance (MGUS). The clinical presentation of MM may be nonspecific, with the most common presenting symptoms being fatigue, bone pain and anaemia. The diagnostic criteria for MM were revised in 2014 to include 3 specific biomarkers of malignancy that are associated with an increased risk of target organ damage. This has resulted in a paradigm shift in the management of MM. The introduction of immunomodulatory agents and proteasome inhibitors has significantly improved the survival of patients with MM. Autologous stem cell transplantation remains the standard of care in younger, fit patients, where there is also a clear role for maintenance chemotherapy. Transplant-ineligible patients benefit from a prolonged induction therapy, and the role of maintenance therapy in this setting is still unclear. Despite major advances in therapy, MM remains an incurable malignant condition and novel agents such as monoclonal antibodies play an important role, especially in the elderly and patients who have relapsed.
Multiple myeloma (MM) belongs to the group of plasma cell neoplasms (Table 1)[1] It is characterised by the neoplastic proliferation of plasma cells that produce an abnormal immunoglobulin or monoclonal (M) protein. MM accounts for 1% of all cancers and ~10% of haematological malignancies in the USA.[2] There is a slight male predominance, with double the number of African Americans affected than whites.[3] The median age at diagnosis is 65 years.[4] The South African (SA) National Cancer Registry indicated 347 new cases of MM for 2014, which accounted for just <1% of all cancers and ~9% of haematological malignancies. The majority of patients were diagnosed in their 70s, with ~18% diagnosed when <50 years of age.[5]
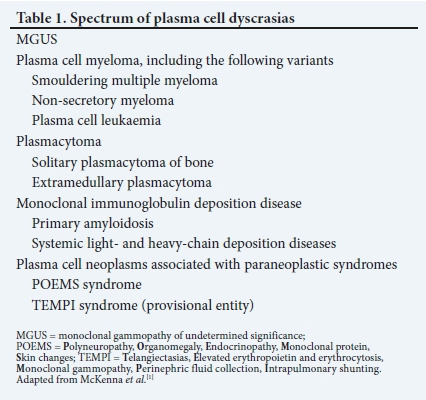
MM is usually preceded by an asymptomatic premalignant condition, i.e. monoclonal gammopathy of undetermined significance (MGUS).[1] MGUS is present in 3 - 4% of people >50 years of age and 5% of people >70 years of age. The risk of progression to MM is 1% per year.[6,7] Smouldering multiple myeloma (SMM) is a more advanced premalignant phase between MGUS and MM. The risk of progression from SMM to MM is 10% per year in the first 5 years, followed by 3% per year in the next 5 years, and 1.5% per year thereafter.[8] This rate of progression is influenced by underlying cytogenetic abnormalities.[9]
The spectrum of presentation of MM varies from asymptomatic to aggressive disease, with the most aggressive presentation being that of a plasma cell leukaemia (PCL).[1]
Clinical presentation
The symptoms of MM are primarily related to bone infiltration by plasma cells or renal tubular damage by excess light chains.[10]
The clinical presentation of MM may be nonspecific. The most common presenting symptoms are fatigue, bone pain and anaemia.[4] Renal failure may be a presenting feature, and about half of newly diagnosed MM patients have a raised creatinine level.[4] Renal insufficiency can be due to hypercalcaemia or a myeloma cast nephropathy caused by increased serum free light chains. Other causes of renal failure include the use of nephrotoxic agents, such as non-steroidal anti-inflammatory drugs (NSAIDs) and secondary light-chain amyloidosis.
Hypercalcaemia may be found in approximately one-third of patients at diagnosis, and may require urgent therapy. While bone pain is present in up to 60% of patients when diagnosed with MM, a third of patients may also present with pathological fractures, usually of the axial skeleton, but also involving the long bones.[4,11]
Occasionally, MM may present as a medical emergency owing to spinal cord compression by an extradural plasmacytoma or vertebral fracture. Furthermore, patients with MM are at increased risk of infections due to impaired cell-mediated and humoral immunity. Infections due to pneumococcal organisms are most common, but those due to Haemophilus, Gram-negative organisms and viruses are also increased.[10,11]
Approximately 7% of patients with MM may present with extra-medullary disease only, which is associated with poorer survival.[12]-Less common clinical presentations include bleeding from platelet dysfunction or coagulation abnormalities, and hyperviscosity symptoms due to a very high M-protein level. Hepatosplenomegaly is unusual, except in cases of amyloid deposition or POEMS (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal protein, Skin changes) syndrome. Some patients with MM may be diagnosed after an incidental finding of a raised globulin fraction on liver function tests performed for other reasons.
Diagnostic approach
Due to the often nonspecific nature of the presenting symptoms of MM, one needs to have a high index of suspicion to make the diagnosis. MM must be considered in the older patient who presents with bone pain, unexplained anaemia, hypercalcaemia or renal failure, for which there is no clear cause.
Clinical evaluation
If a diagnosis of MM is suspected, history taking should pay particular attention to bone pain, constitutional symptoms, infections and neurological complaints. A clinical examination should include assessment for pallor, bone tenderness, spinal deformities and infections. A neurological examination is mandatory, especially with regard to signs of neuropathy and spinal cord compression.[10]
Investigations
Investigations used in the diagnostic evaluation of MM are aimed at demonstrating the M-protein and clonal plasma cells in the bone marrow, and evaluating for end-organ damage. Table 2 summarises some of these investigations.
Blood investigations
Baseline investigations for MM entail a full blood count (FBC) with a differential count and a smear, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP) and biochemical tests, including calcium, urea and electrolytes (U&E), uric acid and liver function tests In our local setting, hepatitis B and C and HIV testing is usually performed.
Common findings on the FBC include a normochromic, normocytic anaemia in up to 73% of patients at diagnosis.[4] More than 50% of patients demonstrate rouleaux formation on the peripheral smear due to high protein levels.[4]Circulating plasma cells in the peripheral blood is an uncommon finding, but if present, may be indicative of a PCL. MM patients who secrete a heavy-chain immunoglobulin have a raised globulin fraction (a high protein with normal-low albumin), while the globulin fraction is usually normal in patients with only light-chain secretion. The majority of patients with MM have decreased levels of uninvolved immunoglobulins (immune paresis), which predispose them to infection.
Screening tests to demonstrate the M-protein include a serum protein electrophoresis (SPEP) with immunofixation, and a serum free light-chain assay or urine protein electrophoresis (UPEP) with immunofixation.[13] More than half of patients with MM produce IgG heavy-chain M-protein, while 20% produce IgA and a similar number secrete only monoclonal light chains (light-chain myeloma). [10] IgD and IgM monoclonal proteins are rare, and the differential diagnosis of a lymphoplasmacytic lymphoma (Waldenstrom macroglobulinaemia) must be considered in a patient with an IgM paraprotein. Patients with light-chain myeloma have a normal SPEP, with light chains detected on the serum free light-chain assay and UPEP. About 2 - 3% of patients have true non-secretory MM with no detectable M-protein in the serum or urine; these patients require imaging and bone marrow biopsy for diagnosis and monitoring.[4,10]
Bone marrow examination
In 2014, the International Myeloma Working Group (IMWG) revised the criteria for the diagnosis of MM and related disorders. Table 3 lists the diagnostic criteria for MM and related plasma cell disorders.[2]
This major revision was to include a subset of patients with previous SMM, who displayed 1 of 3 specific biomarkers in the category of MM.'2- Each biomarker is associated with an 80% 2-year risk of progression to end-organ damage. These biomarkers, together with the CRAB (hyper Calcaemia, Renal failure, Anaemia, Bone lesions) criteria, are known as myeloma-defining events. This revision of the criteria for MM has resulted in a paradigm shift, as a subset of patients with SMM are now treated earlier, prior to developing end-organ damage.
A bone marrow aspirate and trephine biopsy is key to the definitive diagnosis of MM.
Plasma cell clonality is demonstrated on flow cytometry or immunohistochemistry. Fluorescent in situ hybridisation (FISH) studies aid with molecular characterisation of MM, which is important for therapeutic decisions and prognostication (see Risk stratification below).[14]
Imaging
Advanced imaging modalities are more sensitive for the diagnosis of MM and have largely replaced the skeletal survey in the developed world. Imaging techniques include whole-body, low-dose computed tomography (CT) scanning, fluorodeoxyglucose F18 (18F-FDG) positron emission tomography/CT (PET/CT) and magnetic resonance imaging (MRI). PET/CT is more sensitive for soft-tissue lesions, while MRI has superior sensitivity for focal bone marrow involvement.[15] MRI is also useful in cases of suspected spinal cord compression.
A skeletal survey examination (skull, ribs, spine, pelvis, humerus and femur radiographs) is useful in a setting where more specialised imaging is not available. Abnormalities on skeletal survey include punched-out lytic bone lesions, generalised osteopenia and vertebral compression fractures.
Risk stratification
MM is a heterogeneous disease, with the clinical presentation being quite aggressive in some patients, while patients with SMM do not require treatment. The median overall survival of patients with MM is ~6 years. Patient- and disease-related factors affect treatment response and overall survival.[16] Important patient factors include age, performance status and presence of comorbidities, all of which influence the type of induction chemotherapy and transplant eligibility. Disease-related factors include tumour burden and cytogenetics. Staging of MM was previously done according to the Salmon-Durie classification, which involved assessment of tumour burden by degree of anaemia, hypercalcaemia, creatinine, extent of lytic bone disease and immunoglobulin levels.[17] This classification has largely been replaced by the Revised International Staging System (R-ISS) for MM, which incorporates markers of tumour burden, such as albumin and beta-2 microglobulin, as well as indicators of tumour biology, such as lactate dehydrogenase and cytogenetics (Table 4).[18]
The R-ISS is easy to use and more objective than the Salmon-Durie classification.[10]
Management
The standard of care for patients with MGUS and SMM is observation. Treatment is indicated in all patients who fulfil the IMWG criteria for the diagnosis of MM. Without treatment, the median survival is 6 months.[19] The management of MM consists of supportive and specific measures.
Supportive management and adjunctive therapies
The supportive aspect of management is crucial in MM. This is borne out by the observation of newly diagnosed patients entered into the UK's Medical Research Council (MRC) trials, where there was a 10% rate of early death of patients with MM (within 60 days), mainly due to infection, followed by renal failure.[20]
Patients presenting as medical emergency cases require prompt intervention. Urgent radiotherapy is indicated in acute spinal cord compression. Severe hypercalcaemia of rapid onset requires urgent isotonic hydration, steroids and intravenous bisphospho-nate therapy (pamidronate or zoledronic acid).[10] Patients with uncommon presentations of hyperviscosity syndrome need prompt plasmapheresis.
Care should be taken to avoid exacerbation of pre-existing renal impairment. Nephrotoxic drugs such as aminoglycosides and NSAIDs must be avoided, and patients should maintain adequate hydration. Infections must be managed promptly, and patients must receive pneumococcal and influenza vaccines.[10] Appropriate analgesia is important for management of bone pain.
Reversible underlying causes of anaemia must be treated. Patients with acute, symptomatic anaemia require blood transfusion. Erythropoiesis-stimulating agents may be used during the course of the disease, albeit with caution, owing to the increased risk of thrombosis associated with these agents. [10]
Bisphosphonate therapy is integral to the management of MM patients with symptomatic bone disease and lytic lesions or osteopenia on imaging. Apart from treating hypercalcaemia, bisphosphonates improve bone pain, decrease skeletal-related events and have an anti-tumour effect. There is a role for surgery for fixation of pathological or impending fractures of the long bones, as well as spinal cord decompression of vertebral fragments. Vertebroplasty or kyphoplasty are useful adjuncts to pain control. Radiotherapy is also used for intractable bone pain and for the treatment of solitary plasmacytomas.
Principles of specific treatment
Treatment is dictated by transplant eligibility, risk stratification and available resources. Transplant eligibility is determined by age, performance status and comorbidities. In general, fit patients up to the age of 65 - 70 years are considered for transplant. Drug availability due to high costs of newer agents, as well as access to transplantation, is a major challenge in resource-constrained environments.[21]
Transplant-eligible patients
Induction chemotherapy for 3 - 4 cycles followed by high-dose therapy and autologous stem cell transplantation (ASCT) is the standard of care for MM patients who are eligible for transplant. ASCT is not curative, but improves event-free and overall survival compared with chemotherapy alone.[10] Several triplet regimens are recommended, most of which include a proteasome inhibitor, immunomodulatory agent and dexamethasone. Table 5 alludes to the different classes of agents available. Choice of regimen is guided by disease biology, patient characteristics, drug availability and regional guidelines. Some common regimens include bortezomib, lenalidomide and dexamethasone; bortezomib, cyclophosphamide and dexamethasone; and bortezomib, thalidomide and dexamethasone.[81 The pros and cons of the various regimens are beyond the scope of this review. Trials of maintenance therapy after ASCT (bortezomib-based or lenalidomide) have shown improved progression-free and overall survival.[22-24]

Transplant-ineligible patients
Patients who are transplant ineligible may be treated with the same regimens as transplant-eligible patients if they can tolerate these regimens (for 8 - 12 months), but such patients can also receive dual-therapy regimens and melphalan-containing regimens (for up to 18 months).
The role of maintenance in this group of patients is less clear.[8]
Treatment monitoring and relapse
The IMWG uniform response criteria is used to monitor treatment response and relapse of MM.[25] Patients are monitored at clinic visits by SPEP and serum FLC levels or 24-h UPEP. Almost all patients with MM relapse. Management of relapsed/refractory MM is beyond the scope of this article, but there is a clear role for novel agents such as monoclonal antibodies in this setting.
Summary
• MM belongs to the group of plasma cell neoplasms and is characterised by the production of a monoclonal protein (M-protein).
• The most common presenting symptoms of MM are fatigue, anaemia and bone pain.
• One must have a high index of suspicion to diagnose MM in the older patient who presents with nonspecific symptoms.
• Renal impairment is common in MM, and nephrotoxic agents must be avoided in these patients.
• Spinal cord compression, hypercalcaemia and hyperviscosity syndrome are medical emergencies, and should be managed as such.
• A bone marrow aspirate and trephine biopsy are mandatory for the definitive diagnosis of MM.
• The skeletal survey, although largely replaced by modern imaging, still has a useful place in the local setting.
• The supportive management of patients with MM is crucial, and includes maintaining adequate hydration, administering appropriate analgesia and managing concurrent infections.
• Treatment is dictated by transplant eligibility, with ASCT currently being the standard of care in younger, fit patients.
Declaration. None.
Acknowledgements. None.
Author contributions. FF and FB were both involved in the writing of the article, and both approved the final version.
Funding. None.
Conflicts of interest. None.
References
1. McKenna RW, Kyle RA, Kuehl WM, et al. Plasma cell neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th ed. Lyon: International Agency for Research on Cancer, 2017: 241. [ Links ]
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014;15(12):e538-e548. https://doi.org/10.1016/S1470-2045(14)70442-5 [ Links ]
3. Landgren O, Weiss BM. Patterns of monoclonal gammopathy of undetermined significance and multiple myeloma in various ethnic/racial groups: Support for genetic factors in pathogenesis. Leukemia 2009;23(10):1691-1697. https://doi.org/10.1038/leu.2009.134 [ Links ]
4. Kyle RA, Gertz MA, Witzig TE, et al Review of 1 027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003;78(1):21-33. https://doi.org/doi.org/10.4065/78.L21 [ Links ]
5. Cancer Association of South Africa. Statistics. 2014 National Cancer Registry. https://www.cansa.org.za/south-african-cancer-statistics/ (accessed 3 July 2019). [ Links ]
6. Kyle RA, Therneau TM, Rajkumar SV, et al Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 2006;354(13):1362-1369. https://doi.org/10.1056/NEJMc061160 [ Links ]
7. Kyle RA, Larson DR, Therneau TM, et al Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med 2018;378(3):241-249. https://doi.org/10.1056/NEJMoa1709974 [ Links ]
8. Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk-stratification and management. Am J Hematol 2018;93:1091-1110. https://doi.org/10.1002/ajh.25117 [ Links ]
9. Lakshman A, Paul S, Rajkumar SV, et al. Prognostic significance of interphase FISH in monoclonal gammopathy of undetermined significance. Leukemia 2018;32(8):1811-1815. https://doi.org/10.1038/s41375-018-0030-3 [ Links ]
10. Rajkumar SV. Multiple myeloma: Clinical features, laboratory manifestations, and diagnosis. In: Connor RF, ed. UpToDate. Waltham, MA: UpToDate Inc, 2019. https://www.uptodate.com/ (accessed 22 June 2019). [ Links ]
11. Shah D, Seiter K. Multiple myeloma clinical presentation. https://emedicine.medscape.com/ (accessed 22 June 2019). [ Links ]
12. Varettoni M, Corso A, Pica G, et al. Incidence, presenting features and outcome of extramedullary disease in multiple myeloma: A longitudinal study on 1 003 consecutive patients. Ann Oncol 2010;21(2):325. https://doi.org/10.1093/annonc/mdp329 [ Links ]
13. Katzmann JA, Dispenzieri A, Kyle R, et al Elimination of the need for urine studies in the screening algorithm for monoclonal gammopathies by using serum immunofixation and free light chain assays. Mayo Clin Proc 2006;81(12):1575-1578. https://doi.org/10.4065/81.12.1575 [ Links ]
14. Rajan AM, Rajkumar SV Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J 2015;5:e365. https://doi.org/10.1038/bcj.2015.92 [ Links ]
15. Zamagni E, Nanni C, Patriarca F, et al. A prospective comparison of 18F-fluorodeoxyglucose positron emission tomography-computed tomography, magnetic resonance imaging and whole-body planar radiographs in the assessment of bone disease in newly diagnosed multiple myeloma. Haematologica 2007;92(1):50. https://doi.org/10.3324/haematol.10554 [ Links ]
16. Russell SJ, Rajkumar SV. Multiple myeloma and the road to personalised medicine. Lancet Oncol 2011;12(7):617-619. https://doi.org/10.1016/S1470-2045(11)70143-7 [ Links ]
17. Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation ofmeasured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer 1975;36:842-854. https://doi.org/10.1002/1097-0142(197509)36:3<842::aid-cncr2820360303>3.0.co;2-u [ Links ]
18. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A report from International Myeloma Working Group. J Clin Oncol 2015;33(26):2863-2869. https://doi.org/10.1200/JCO.2015.61.2267 [ Links ]
19. Osgood EE. The survival time of patients with plasmocytic myeloma. Cancer Chemother Rep 1960;9:1. [ Links ]
20. Augustson BM, Begum G, Dunn JA, et al. Early mortality after diagnosis of multiple myeloma: Analysis of patients entered onto the United Kingdom Medical Research Council trials between 1980 and 2002 - Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 2005;23(36):9219. https://doi.org/10.1200/JCO.2005.03.2086 [ Links ]
21. Rajkumar SV. Multiple myeloma: Management in resource-poor settings. Connor RF, ed. UpToDate. Waltham, MA: UpToDate Inc, 2019. https://www.uptodate.com/ (accessed 26 June 2019). [ Links ]
22. Sonneveld P, Schmidt-Wolf IG, van der Holt B, et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: Results of the randomized phase III HOVON-65/GMMGHD4 trial. J Clin Oncol 2012;30(24):2946-2955. https://doi.org/10.1200/JCO.2011.39.6820 [ Links ]
23. Moreau P, Attal M, Facon T. Frontline therapy of multiple myeloma. Blood 2015;125:3076-3084. https://doi.org/10.1182/blood-2014-09-568915 [ Links ]
24. Attal M, Palumbo A, Holstein SA, et al. Lenalidomide maintenance after high-dose melphalan and autologous stem-cell transplantation in multiple myeloma: A meta-analysis of overall survival. J Clin Oncol 2016;34(16 Suppl):8001. https://doi.org/10.1200/JCO.2016.34.15_suppl8001 [ Links ]
25. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016;17(8):e328-e346. https://doi.org/10.1016/S1470-2045(16)30206-6 [ Links ]
 Correspondence:
Correspondence:
F Fazel
fbfazel@gmail.com
Accepted 26 August 2019


{kind=link}
{kind=link}
{kind=link}