










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.109 no.9 Pretoria sep. 2019
http://dx.doi.org/10.7196/samj.2019.v109i9.13936
RESEARCH
Role of family history and clinical screening in the identification of families with idiopathic dilated cardiomyopathy in Johannesburg, South Africa
C BaillyI; S HenriquesII; N TsabedzeIII; A KrauseIV
IMB ChB, DCH, FCMG (SA), MMed (Medical Genetics); Division of Human Genetics, National Health Laboratory Service; and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIBSc Hons Biological Sciences, MSc Science Communication, MSc Genetic Counselling; Division of Human Genetics, National Health Laboratory Service; and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIMB BCh, FCP (SA), Cert Cardiology (SA); Division of Cardiology, Department of Internal Medicine, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVMB BCh, PhD; Division of Human Genetics, National Health Laboratory Service; and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND. Familial disease is implicated in 20 - 50% of cases of idiopathic dilated cardiomyopathy (IDCM) worldwide. The contribution of familial factors to IDCM in the Johannesburg area, South Africa, is unknown.
OBJECTIVES. To describe the demographic details of patients with IDCM who presented at Charlotte Maxeke Johannesburg Academic Hospital (CMJAH), and to determine if there is evidence of familial disease through family history assessment and clinical screening of relatives.
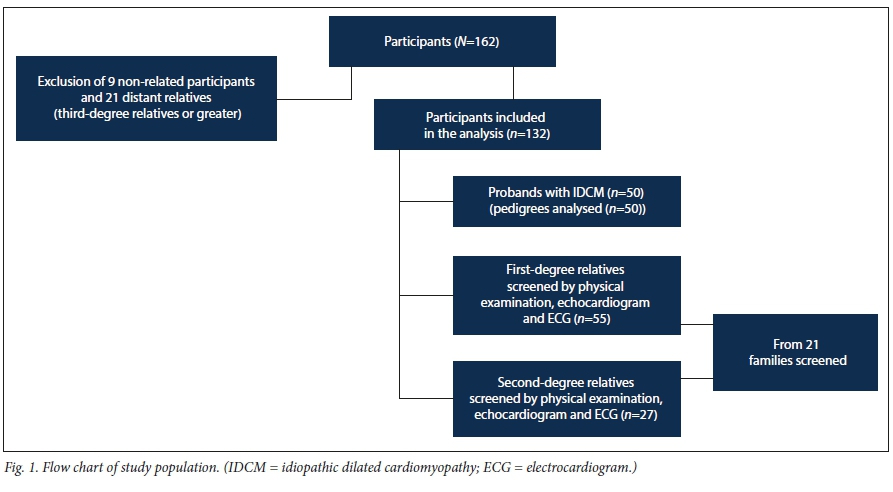
METHODS. This was a single-centre, cohort study performed at a quaternary care centre at CMJAH. Fifty unrelated probands diagnosed with IDCM and available first- and second-degree relatives were included in the study. A three-generation family pedigree was drawn up for all 50 probands. The pedigrees were analysed to identify the presence or absence of familial disease and categorised as positive, intermediate, negative or unreliable according to the family history obtained. From the 50 proband cases, there were 21 family members available for screening for features of IDCM. Eighty-two family members (55 first-degree and 27 second-degree relatives) were screened clinically. Screening included a personal history, full physical examination, electrocardiogram (ECG) and echocardiogram.
RESULTS. The mean age at diagnosis of IDCM in the probands was 41.7 (standard deviation (SD) 12.4) years. The majority of probands were males (n=38; 76%). Of 50 pedigrees analysed, 14 (28%) were positive and likely to be indicative of familial dilated cardiomyopathy (DCM), and 9 (18%) patients were at intermediate risk of familial disease. Eighty-two asymptomatic family members were screened, with a median age of 33 (range 11 - 76) years. No asymptomatic family members were identified with features of DCM or presymptomatic DCM. Eleven of the 21 families screened had relatives with possible presymptomatic DCM identified by abnormalities on the echocardiogram in 3 families (14.3%) (4 individuals; all first-degree relatives of the index case) or identified on the basis of a conduction defect (an arrhythmia or first-/ second-/third-degree heart block) in 8 families (72.7%) (11 individuals; 9 first-degree and 2 second-degree relatives).
CONCLUSIONS. Screening for IDCM should include a three-generation family history and clinical screening of all first-degree family members. As IDCM has an age-related penetrance, at-risk family members should receive follow-up for screening to assess symptoms and signs of IDCM. Genetic testing would potentially identify family members at high risk, who would benefit from screening; this might be a less expensive option.
Cardiac failure is the most common cardiovascular manifestation in urban Africa[1] Dilated cardiomyopathy (DCM), defined as left ventricular (LV) dilatation and systolic dysfunction, is a major cause of heart failure in adults between the third and fourth decades of life[1-3] It is the leading cause for heart transplantation worldwide.[3] Although population-based data on the burden of DCM in sub-Saharan Africa are lacking, it is reported to account for up to 48% of patients hospitalised with cardiac failure. Idiopathic dilated cardiomyopathy (IDCM) was the second most common form of cardiomyopathy after hypertensive cardiomyopathy in the Heart of Soweto Study Cohort[6]
IDCM, a form of DCM without an identifiable cause, can be diagnosed after exclusion of secondary causes and other primary cardiomyopathies. Secondary causes include untreated hypertension, myocarditis triggered by infection, coronary artery disease, valvular heart disease, congenital heart disease, autoimmune disease, metabolic factors, alcohol abuse and nutritional deficiencies.t1,41 The other primary cardiomyopathies, which are classified according to morphofunctional phenotype, include hypertrophic obstructive cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy (ARVC) and LV non-compaction cardiomyopathy (LVNCC)[7] Genetic conditions with dilated cardiomyopathy as a presenting feature include the muscular dystrophies (Duchenne and Becker muscular dystrophy, as well as their carrier states, Emery-Dreifuss muscular dystrophy and limb-girdle muscular dystrophy), hereditary haemochromatosis, Friedreich's ataxia, Barth syndrome, mitochondrial myopathies and numerous inborn errors of metabolism.[8,9] Up to half of all cases of IDCM are believed to be hereditary or familia[1,2,4,10] Timeous referral of DCM patients for management of cardiac failure, arrhythmias and life-saving interventions such as cardiac transplantation is of the utmost importance. The abovementioned factors highlight the significance of identifying patients with familial DCM and screening their relatives to identify at-risk presymptomatic family members.[11]
Patients with DCM most commonly present with decompensated cardiac failure.[10] Other manifestations may include arrhythmias, sudden cardiac death and, less commonly, a thromboembolic eventJ8,11,12] Most patients with DCM are diagnosed when they become symptomatic; however, presymptomatic affected individuals can be detected during specialist cardiovascular examination by identifying echocardiographic signs of LV enlargement, a decreased ejection fraction and/or fractional shortening, wall motion abnormalities and atrial enlargement, and electrocardiographic features, including a primary, secondary or tertiary atrioventricular block, a left or right bundle branch block, abnormal QRS patterns (including left or right axis deviation), premature ventricular or atrial contractions, atrial fibrillation or flutter and ventricular arrhythmias/ tachycardias.!81 This so-called presymptomatic stage may persist tor months to years without the onset of symptoms.!81 Identifying presymptomatic individuals can provide an opportunity tor invoking lifestyle changes and allow for pharmacological therapy to be initiated in the earlier stages of the course of the disease, with the aim of limiting the progression of cardiac failure and controlling arrhythmia.[10,31] Diagnosis of DCM requires specialist investigations such as echocardiography, which is mostly limited to tertiary medical centres.[4]
Familial DCM is a monogenic disorder with mutations identified in >40 genes. It is mostly inherited in an autosomal dominant manner, although autosomal recessive, X-linked and mitochondrial patterns of inheritance have been described.[2, 14] Autosomal dominant familial DCM is characterised by incomplete and/or variable expressivity with regard to age of onset, severity of symptoms and risk of complications.[14] Advances in genetic testing have changed the approach to genetic diagnoses of the cardiomyopathies. Familial DCM is the most genetically heterogeneous of the cardiomyopathies.[15] a major role-player in familial DCM is the TTN gene, encoding the protein titin. Approximately 20 - 25% of familial DCM patients have a truncating mutation in this gene[15] Truncating mutations in the TTN gene have also been found in patients with peripartum cardiomyopathy, which shares clinical features with IDCM and may be part of the spectrum of familial DCM.[16,17] In some patients with familial DCM, the findings of cardiac conduction defects may point to a mutation in certain genes such as LMNA and SCN5A. Identifying the genetic mutation may alter specific management. For example, it is recommended that individuals with LMNA mutations benefit from early implantable cardioverter defibrillators (ICDs)J[819] Targeted gene panels using next-generation sequencing (NGS) are commonly used in developed countries and have a diagnostic yield of 30 - 35%.[9,19,10]' The causative mutations in South African (SA) patients have not been investigated or identified to date.
Objectives
The aim of this article was to describe the demographic details of patients with IDCM in the Johannesburg area and to determine, by family history assessment and clinical screening of relatives, if evidence of familial disease exists.
Methods
Study design and study population
This was a single-centre study performed at the Department of Cardiology, Charlotte Maxeke Johannesburg Academic Hospital (CMJAH).
Each proband diagnosed with IDCM had to fulfil all of the following criteria: (z) clinical evidence of cardiac failure with LV dilatation; (ii) LV ejection fraction (LVEF) of <50% on echocardiography; and (Hi) exclusion of common secondary causes of DCM. All probands were offered clinical screening for tamily members; however, only 21 of the probands' family members were available for screening (Fig. 1).
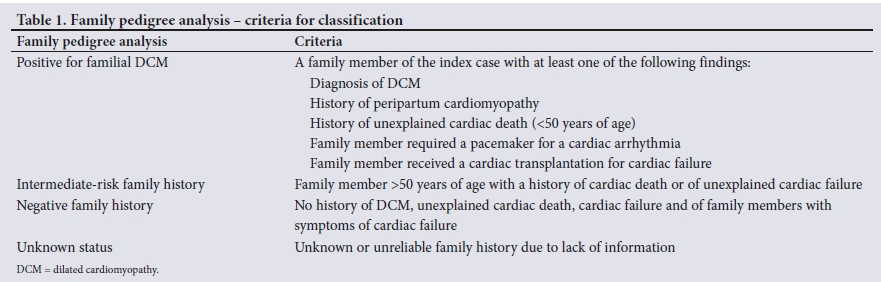
All study participants had undergone a comprehensive clinical assessment, including a three- to four-generation family pedigree, personal medical history, clinical examination (performed by a cardiologist (NT)), electrocardiogram (ECG) and echocardiogram. Data collected for each study participant included ethnicity, gender, current age, age at which the proband was diagnosed with IDCM, clinical cardiovascular examination detail, ECG and echocardiogram findings. Further relevant family history included ischaemic heart disease, cerebrovascular disease that possibly reflected a thromboembolic event, hypertension or other relevant genetic conditions, including muscular dystrophies. Based on family history, a pedigree was constructed and analysed. Each family was classified as either positive for familial DCM, having an intermediate-risk or negative family history or unknown status. The criteria are set out in Table 1. Patient accounts of cardiovascular diagnoses and their aetiologies are not always reliable, cannot necessarily be proven and need to be interpreted with cautionJ8! When an individual with IDCM gives a history of family members with heart failure, sudden death, heart attacks, arrhythmias, pacemakers or heart failure symptoms, such a history should not be ignored, but should raise suspicion of familial cardiac disease, including familial DCM, especially when the relatives are young[8] This was the rationale for the criteria used to classify the family pedigrees during analysis.
Screening of asymptomatic family members included a physical examination, ECG and echocardiography. Echocardiography assessment included LV internal diameter in diastole (LVIDd) and LVEF. ECG screening was performed for cardiac conduction abnormalities, including a primary, secondary or tertiary atrioventricular block, a left or right bundle branch block, abnormal QRS patterns (including left or right axis deviation), premature ventricular or atrial contractions, atrial fibrillation or flutter and ventricular arrhythmias/tachycardias. The echocardiogram and ECG findings were interpreted with assistance from NT.
Based on the abovementioned results, each asymptomatic family member was categorised into one the following groups: (i) features of DCM; (ii) features of presymptomatic DCM; (iii) possible presymptomatic DCM and cardiac conduction abnormality; and (iv) no features of DCM. Criteria for categorisation of family members based on screening are set out in Table 2.
Ethical approval
The study was approved by the University of the Witwatersrand Human Research Ethics Committee (ref. no. HREC M150467). The study participants were recruited from the co-author's (NT) study entitled 'Genetics of idiopathic dilated cardiomyopathy in Johannesburg' and included the first 50 probands diagnosed with IDCM and available family members seen over an 18-month period (July 2015 - February 2017). Written informed consent was obtained by the co-authors for the utilisation of relevant clinical data.
Statistical analysis
Statistical analysis was performed using TIBCO Statistica 13.3.0 (TIBCO Software Inc., USA). Descriptive statistics of the study cohort were applied as either means with standard deviations (SDs) if normally distributed or medians with interquartile ranges if not normally distributed. Frequency analysis was performed for discrete variables and represented as percentages, with 95% confidence intervals (CIs) where appropriate. A f-test was performed to assess for significant age differences between male and female probands. A p-value of <0.05 was considered statistically significant.
Results
Index case demographics
Fifty probands were included in the study cohort. Of these, 38 (76%) were males. The mean age at diagnosis of IDCM was 41.7 (SD 12.4) years for all probands, 42.5 (SD 11.24) years for males and 39.3 (SD 15.85) years for females, with no statistically significant differences in the ages of men and women (p=0.5). The racial distribution comprised 43 (86%) black Africans, 3 (6%) whites, 3 (6%) of mixed ancestry and 1 (2%) of Indian descent.
Pedigree analysis
As per the criteria set out in Table 1, 14 (28.0%) of the 50 pedigrees analysed had a positive family history of familial DCM, 9 (18%) an intermediate-risk family history, 12 (24%) a negative family history and 15 (30%) an unreliable family history. Positive or intermediate family pedigrees were therefore seen in 23 (46%) families. A summary of the pedigree analysis can be seen in Fig. 2.
Family member analysis
Fifty-five first-degree relatives were screened, including 25 (45.5%) males and 30 (54.5%) females, with a median age of 38 (range 11 -76) years. Twenty-seven second-degree relatives were screened, including 11 (41%) males and 16 (59%) females, with a median age of 22 (range 12 - 72) years.
According to the criteria stated in the abovementioned methods section, none of the 82 asymptomatic family members included in the screening analysis had identifiable features of DCM. One first-degree family member who attended a screening had already been diagnosed with IDCM and was symptomatic. None of the 82 asymptomatic family members were identified with presymptomatic DCM.
Eleven families with members with possible presymptomatic DCM were identified. In 3 (14%) families, 4 first-degree relatives of the probands had abnormalities detected on echocardiography. In 8 (38%) families, 11 individuals were identified with possible presymptomatic DCM, based on finding a cardiac conduction defect (either a sinus node dysfunction or a first-/second-/third-degree heart block). Nine (81.8%) of the 11 individuals were first-degree relatives and 2 (18.2%) were second-degree relatives. No identifiable features of DCM were detected in 10 (48%) families screened. The screening results for the asymptomatic family members are summarised in Table 3.
From the 15 individuals with possible presymptomatic DCM, 4 (26.7%) were from 4 families with a positive family history, 6 (40%) were identified from 3 families with an intermediate family history, 3 (20%) were identified in 2 families with a negative history and 2 (13.3%) were identified in 2 families with an unknown family history. Ten (66.7%) individuals with possible presymptomatic DCM were therefore identified in high-risk families (i.e. those with positive and intermediate-risk family histories).
Discussion
Pedigree analysis
IDCM is associated with a high mortality and is an important contributor to the burden of disease in Africa.!1! This study shows that patients with IDCM seen at CMJAH are diagnosed at a similar age as those reported elsewhere, with an average age of diagnosis of 41 years. Our study also shows that a greater percentage of males appear to be affected than females, with 76% of index cases being males. This is in accordance with the literature, where males are almost twice as likely to be affected.[1-4]
A positive family history was found in 14 (28%) of the 50 family pedigrees analysed, which is similar to findings reported by Ntusi et al,[5] where familial disease was found in 26.6% of patients with IDCM by family pedigree analysis in Cape Town. The pedigrees with positive and intermediate family histories appeared most consistent with an autosomal dominant pattern of inheritance: 13 of 14 (93%) positive family histories were clearly consistent with an autosomal dominant inheritance pattern and only 1 of 14 (7%) had a possible autosomal recessive or X-linked inheritance pattern - a family with male non-identical twins affected with IDCM. This may also be explained by a germline mutation in a parent or autosomal dominant disease with reduced penetrance. The interpretation of the family histories may be further complicated by non-penetrance, age-related penetrance and variable expressivity, which may be features of familial DCM and appeared to be evident in the family pedigrees analysed. In only 4 of 14 (26%) cases with a positive family history did the proband give a history of a relative with a known diagnosis of IDCM.
In 2 of 9 (22%) intermediate-risk families, there was a history of unexplained cardiac failure, emphasising the importance of a diagnosis of the cause of cardiac failure and exclusion of secondary causes and other primary cardiomyopathies. The remainder of the intermediate-risk families (7 of 9; 77%) had unexplained death believed to be due to a cardiac event in family members >50 years of age. This may illustrate the possible age-related penetrance and variable expressivity of familial DCM in individuals who died; however, aetiologies of these deaths are unknown and may also be due to non-cardiac causes or cardiac causes unrelated to familial DCM. A high index of suspicion is needed if there is a family history of any individual with symptoms of cardiac failure, arrhythmias or unexplained sudden death.!8! Symptoms of typical cardiac failure are nonspecific, which is a limitation to the validity and accuracy of a family history, further emphasising the importance of an objective evaluation of LV function, particularly in first-degree relatives of probands with IDCM.
Twelve (24%) of the probands who were classified as having a negative family history were able to give a detailed family history where there was no history of any relatives with cardiac disease or unexplained cardiac deaths, and the proband appeared to be the first and only case of IDCM in the pedigree. This occurrence of a negative family history appears to be much lower than that reported by Ntusi et at.,151where 73.4% of their cases of IDCM appeared to be non-familial. This can be explained by our classification of the family pedigrees, which included intermediate-risk family histories and unknown or unreliable family histories, resulting in a lower negative family history.
An unknown or unreliable family history was found in 15 (30%) cases. This was mostly explained by probands not having recent contact with relatives who resided in different provinces or countries, not having knowledge of the cause of death of relatives, difficulty in accessing death certificates of relatives with unexpected or unknown death, and a general lack of knowledge of their relatives and family histories. These factors make a family pedigree as a primary assessment tool somewhat insensitive. A positive family history is significant and requires follow-up, but a negative family history does not exclude familial DCM.
Screening analysis
In a single family, 1 individual (a sibling of the index case) who attended screening was symptomatic and had been previously diagnosed with IDCM. No family members screened fulfilled criteria for features of DCM or presymptomatic DCM. This may be explained by the disease penetrance typically occurring in adult life, while relatively young adults (median 33 (range 11 - 76) years) were screened, who may not yet have developed detectable cardiac abnormalities.
In 11 of 21 (52.4%) families, criteria for possible presymptomatic DCM were identified. The condition was identified by an abnormality on the echocardiogram in 3 (14.3%) families - all first-degree relatives of the index case. The echocardiographic abnormalities identified included an increased LVIDd in 3 individuals (2 from the same family) and a reduced LVEF in 1 individual. None of the 27 second-degree relatives had abnormalities detected on echocardiography, showing that such screening appears more beneficial for first-degree relatives, as might be predicted.
Eight of 21 (38.1%) families with possible presymptomatic DCM were identified based on a cardiac conduction defect (either a sinus dysfunction or first-/second-/third-degree heart block) in a family member. Due to limited specificity of cardiac failure symptoms, we opted to screen all available family members to objectively confirm or exclude DCM. This led to a high number of second-degree relatives being screened. Screening should ideally be limited to first-degree relatives; screening of second-degree relatives is usually only performed when there are anxious relatives or in families with a particularly lethal or penetrant phenotype. [8, 19] Ten of 15 (66.7%) individuals with possible presymptomatic DCM were found in the positive and intermediate-risk family pedigrees, showing the importance of family history in identifying relatives for screening.
SA patients suffering from IDCM often live in resource-limited environments, where there may be a delay in diagnosis. When a patient is diagnosed with IDCM, there is a good opportunity for screening family members and patient education regarding the possibility of the condition being familial.
An evaluation of patients with IDCM should include investigating whether the disease is familial.[19] A cascade approach to screening of first-degree relatives should be used including:
• taking a three-generation family history, keeping in mind the lack of specificity of cardiac failure symptoms and the possibility of unknown and unreliable family histories
• clinical screening for cardiomyopathy in first-degree relatives through clinical examination, an echocardiogram and an ECG to determine if any relatives are affected.[2,9]
The landscape of genetic testing is changing with the advent of NGS technology. As whole exome or genome genetic testing becomes more accessible, cardiac clinicians will increasingly request genetic testing for probands and family members. The role of the geneticist will be for pre- and post-test counselling, patient education and interpreting results of genetic testing. Specialty centres will need to be established for these patients. Genetic testing for the cardiomyopathies, including IDCM, is not available for the majority of patients in SA who make use of State genetic services, but limited testing is available in the private sector. Genetic testing may be offered to individuals with sporadic IDCM, familial DCM or peripartum and pregnancy-associated cardiomyopathy.[9] The rationale for identifying a causal mutation would be to allow mutation-specific cascade screening of family members. This will determine which family members require/ do not require ongoing surveillance.[2,15] When a DCM mutation is identified in an asymptomatic individual, annual screening by physical examination, echocardiography and ECG is recommended to commence from childhood. When a familial mutation has not been identified or testing has not been performed, first-degree relatives of an individual with IDCM should be screened every 3 - 5 years from childhood.[15,18,19] Limitations to genetic testing would include the high cost and possible low detection rate of pathogenic mutations. Mutations may be population specific, and distinguishing clearly pathogenic mutations from benign polymorphisms may be a challenge, as the genetics of IDCM has not been fully investigated and established in African, including SA, patients. These genetic studies need to be done, and once local mutation profiles are established, may provide an effective way to identify presymptomatic at-risk family members.
Study limitations
Our study has some limitations. Patients are often uncertain of their family history and uninformed regarding the cause of death of their relatives, which is a major limitation to interpreting a family history. Furthermore, reviewing hospital records of deceased relatives is a challenge when the relatives died in peripheral hospitals. The small number of first-degree relatives screened is a limitation, as most guidelines advocate for screening of first-degree relatives only in IDCM. In our setting, this was difficult, as many relatives lived far away, some even in neighbouring countries, and were unable to attend screening. This would possibly also have affected the accuracy of the family histories, as many individuals had not had recent and frequent contact with family members. The small number of probands included in the study possibly influenced the capacity to detect significant findings.
Conclusions
Our research has shown that familial disease is common in patients presenting with IDCM at CMJAH. Forty-sixty percent of probands had a positive or intermediate-risk family history. Family pedigrees were almost exclusively consistent with an autosomal dominant inheritance pattern, showing variable expressivity and age-related penetrance, with probands giving family histories of cardiac disease and deaths in individuals <50 years and >50 years of age. Screening for familial disease with history alone is insufficient, as an unreliable and unknown family history was found in 30% of cases.
We have also shown the benefit of screening first-degree family members. No asymptomatic family members screened by ECG and echocardiogram were found to have features of DCM or presymptomatic DCM. Relatives were identified in 11 of 21 (52.4%) families with possible presymptomatic DCM with cardiac conduction abnormality. Co-ordinating the family screening to include first-degree relatives appears to be appropriate and is best undertaken with the assistance of a professional trained in genetics, who can interpret complicated family histories and pedigrees.
These findings have an impact on the clinical evaluation of patients with IDCM at CMJAH. The evaluation of a proband should include a thorough three-generation family history, genetic counselling for the family, and 3 - 5-yearly clinical screening by physical examination, echocardiogram and ECG of first-degree relatives. Although the age of commencement of screening is uncertain, guidelines suggest screening from childhood, especially in cases of early onset and aggressive phenotypes. This represents a significant challenge and cost in our current healthcare system, particularly if first-degree relatives live significant distances from centres with cardiology services. Future research should include identifying the causative genetic mutations that contribute significantly to IDCM in SA and establishing locally appropriate genetic testing. Patients presenting with IDCM could then be given accurate genetic counselling and testing, and screening could be rationalised to high-risk family members who are carriers of a predisposing mutation.
Declaration. This research was conducted by CB in partial fulfilment of his MMed degree in medical genetics.
Acknowledgements. Our sincere appreciation goes to all of the 'Genetics of idiopathic dilated cardiomyopathy in Johannesburg' study investigators. We are grateful to the patients and their family members who participated. CB thanks Profs A Krause and N Tsabedze for their support and guidance throughout the study.
Author contributions. NT, AK, SH conceived the study topic. CB developedand wrote the study protocol, was involved in the recruitment and screening of the participants, collected and analysed the pedigree data and wrote the article. NT and AK supervised the study and data collection, revised the article and approved the final version.
Funding. None.
Conflicts of interest. None.
References
1. Ntusi NB, Badri M, Gumedze F, Wonkam A, Mayosi BM. Clinical characteristics and outcomes of familial and idiopathic dilated cardiomyopathy in Cape Town: A comparative study of 120 cases followed up over 14 years. S Afr Med J 2011;101(6):399-404. [ Links ]
2. Park HY. Hereditary dilated cardiomyopathy: Recent advances in genetic diagnostics. Korean Circ J 2017;47(3):291-298. https://doi.org/10.4070/kcj.2016.0017 [ Links ]
3. Tayal U, Prasad S, Cook SA Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med 2017;9(1):20. https://doi.org/10.1186/s13073-017-0410-8 [ Links ]
4. Sliwa K, Damasceno A, Mayosi BM. Epidemiology and etiology of cardiomyopathy in Africa. Circulation 2005;112(23):3577-3583. https://doi.org/10.1161/CIRCULATI0NAHA.105.542894 [ Links ]
5. Ntusi NB, Wonkam A, Shaboodien G, Badri M, Mayosi BM. Frequency and clinical genetics of familial dilated cardiomyopathy in Cape Town: Implications for the evaluation of patients with unexplained cardiomyopathy. S Afr Med J 2011;101(6):394-398. [ Links ]
6. Stewart S, Wilkinson D, Hansen C, et al. Predominance of heart failure in the Heart of Soweto Study cohort: Emerging challenges for urban African communities. Circulation 2008;118(23):2360-2367. https://doi.org/10.1161/CIRCULATIONAHA.108.786244 [ Links ]
7. Arbustini E, Narula N, Tavazzi L, et al. The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol 2014;64(3):304-318. https://doi.org/10.1016/j.jacc.2014.05.027 [ Links ]
8. Hanson EL, Hershberger RE. Genetic counseling and screening issues in familial dilated cardiomyopathy. J Genet Couns 2001;10(5):397-415. https://doi.org/10.1023/A:1016641504606 [ Links ]
9. Hershberger RE, Siegfried JD. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2011;57(16):1641-1649. https://doi.org/10.1016/j.jacc.2011.01.015 [ Links ]
10. Broch K, Andreassen AK, Hopp E, et al. Results of comprehensive diagnostic work-up in 'idiopathic' dilated cardiomyopathy. Open Heart 2015;2(1):e000271. https://doi.org/10.1136/openhrt-2015-000271 [ Links ]
11. Mestroni L, Maisch B, McKenna WJ, et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J 1999;20(2):93-102. [ Links ]
12. Mestroni L, Rocco C, Gregori D, et al. Familial dilated cardiomyopathy: Evidence for genetic and phenotypic heterogeneity. J Am Coll Cardiol 1999;34(1):181-190. [ Links ]
13. Taylor MR, Carniel E, Mestroni L. Cardiomyopathy, familial dilated. Orphanet J Rare Dis 2006;1:27. https://doi.org/10.1186/1750-1172-1-27 [ Links ]
14. Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010;31(22):2715-2726. https://doi.org/10.1093/eurheartj/ehq271 [ Links ]
15. McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab 2015;21(2):174-182. https://doi.org/10.1016/j.cmet.2015.01.013 [ Links ]
16. Tibazarwa K, Sliwa K, Wonkam A, Mayosi BM. Peripartum cardiomyopathy and familial dilated cardiomyopathy: A tale of two cases. Cardiovasc J Afr 2013;24(5):e4-e7. https://doi.org/10.5830/CVJA-2013-027 [ Links ]
17. Ware JS, Li J, Mazaika E, Yasso CM, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 2016;374(3):233-241. https://doi.org/10.1056/NEJMoa1505517 [ Links ]
18. Skrzynia C, Berg JS, Willis MS, Jensen BC. Genetics and heart failure: A concise guide for the clinician. Curr Cardiol Rev 2015;11(1):10-17. [ Links ]
19. Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol 2016;67(25):2996-3010. https://doi.org/10.1016/j.jacc.2016.03.590 [ Links ]
20. Garcia-Pavia P, Cobo-Marcos M, Guzzo-Merello G, et al. Genetics in dilated cardiomyopathy. Biomark Med 2013;7(4):517-533. https://doi.org/10.2217/bmm.13.77 [ Links ]
 Correspondence:
Correspondence:
C Bailly
claudedidierbaill-y@gmail.com
Accepted 3 April 2019


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}