










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.109 n.1 Pretoria Jan. 2019
http://dx.doi.org/10.7196/samj.2019.v109i1.13415
IN PRACTICE
CLINICAL UPDATE
Cystic fibrosis: What's new in South Africa in 2019
M Zampoli
MB BCh, DCH, FCPaed, Cert Paed Puim (SA); on behalf of the South African Cystic Fibrosis Medical and Scientific Advisory Committee (MSAC); Cystic Fibrosis Clinic, Red Cross War Memorial Children's Hospital, Cape Town, South Africa; and Division ofPaediatric Pulmonology Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
Cystic fibrosis (CF) is one of the most common autosomal recessive disorders worldwide. The incidence of CF depends on the prevalence of CFTR (cystic fibrosis transmembrane conductance regulator) gene mutations in the population, which is determined by genetic diversity and ethnicity. Over 2 000 CFTR mutation variants, divided into six distinct functional classes, are now identified, but not all cause CF disease. Advancements in the molecular diagnosis of CF and recognition of a wider spectrum of CF severity have led to recent revision of CF diagnostic nomenclature and criteria. Identifying which CFTR mutations people with CF carry is important, as novel treatments that target the specific CFTR dysfunction at a molecular level are now available, and many new drugs are in the pipeline. These and other advancements in CF are comprehensively covered in the revised 5th edition of the South African Cystic Fibrosis Consensus Guidelines, published in 2017. In addition, the South African Cystic Fibrosis Registry Initiative (SACFRI) was launched in April 2018. SACFRI is a multicentre public-private collaboration of CF healthcare providers across South Africa (SA), which will prospectively collect data relating to CF diagnosis and outcomes in SA. Local SA registry data are critical to understanding the epidemiology of CF in SA, and SACFRI will be an important tool to identify and prioritise areas of CF care that require intervention.
Cystic fibrosis (CF) is a serious autosomal recessive disorder that occurs with varying frequency in all population groups throughout the world, including South Africa (SA). Although CF survival has improved over the past two decades, it remains a life-shortening condition with median survival age now reported in the USA to be ~40 years. [1]Despite early pioneering work relating to the molecular diagnosis of CF in SA,[2]little is known of the current prevalence, clinical spectrum and outcomes of CF in the SA population, which has wide ethnic and genetic diversity. Data from 2005 from the Red Cross War Memorial Children's Hospital CF clinic cohort in Cape Town indicated that the median age of diagnosis and survival age during the preceding 33 years were 6 months and 19.8 years, respectively.[3]Absence of newborn screening, limited access to sweat testing for diagnosis and restricted availability of expensive CF medications (such as inhaled rhDNAse) or lung transplantation are barriers to improving CF survival in SA. Early and accurate diagnosis and understanding the epidemiology of CF in SA are important to improve outcomes and inform health intervention policies.
In order to address the limitations in CF diagnostic capacity and care in SA, the South African Cystic Fibrosis Medical and Scientific Advisory Committee (MSAC) wishes to draw the attention of the readership of the SAM] to a few important local and international advancements in the field of CF.
The South African Cystic Fibrosis Consensus Guidelines: 5th revision, 2017
The MSAC, in partnership with the South African Cystic Fibrosis Association (SACFA), has revised and published the 5th edition of these guidelines, which comprehensively cover all aspects of CF diagnosis and management appropriate to the SA setting. The guidelines are freely available on the SACFA website (http://www.sacfa.org.za/wp-content/uploads/2017_09_14_CF_Consensus_Guidelines_2017.pdf).[4]We wish to highlight a few important new concepts that appear in this latest revision.

Diagnosis of CF
Molecular diagnosis
The diagnosis of classic or typical CF is confirmed by a positive sweat test (sweat chloride >60 mmol/L) and/or the identification of two disease-causing CFTR (cystic fibrosis transmembrane conductance regulator) mutations on DNA analysis. Over 2 000 CFTR mutations, now classified into six distinct functional classes, have been described to date, of which -200 are classified as 'disease causing'. The Cystic Fibrosis Foundation's updated consensus statement on CF diagnosis recommends the following broad classification of CFTR mutations and has adopted the CFTR2 database (https://www.cftr2.org/index.php) as its reference;[5]
-
CF-causing mutation: results in CF when two abnormal alleles are present in an individual
-
Mutation of varying clinical consequence (MVCC): a mutation that in combination with a CF-causing mutation or another MVCC may result in CF
-
Mutation of unknown clinical consequence: not evaluated by CFTR2
-
Non-CF-causing mutation.
The recognition that the inheritance of any combination of CFTR mutations will result in a wide spectrum of CF phenotypes is important. Healthcare providers are advised to refer to the CFTR2 database to establish the clinical significance of CFTR mutations that are identified. Consultation with a CF expert and a genetic counsellor is strongly recommended when a CFTR mutation is identified.
Commercially available CFTR genotype panels available in SA can detect up to 50 known CFTR mutations prevalent in the SA population. Included in this panel is p.Phe508del (previously known as AF508), the most common mutation in Caucasian and mixed-race people, and c.2988+lG>A (previously known as 3120+1G>A), the most common mutation in black African people. This approach is unsuitable for the SA setting, however, as 79% of black Africans, 55% of individuals of mixed race and 17% of Caucasians will need further molecular testing to confirm a diagnosis of CF.[2]The high proportion of unknown mutations, especially in non-Caucasians with CF, reflects the ethnic and genetic diversity in the SA population. There are few data from the African continent, but a survey identifying disease-causing CF mutations in people of African descent in the Americas found that 55% of alleles still remained unidentified. [6]
Next-generation sequencing (NGS) technology, which screens the whole CFTR gene for abnormal alleles, is becoming more accessible and affordable in SA. Although much more powerful than genotyping for detecting CFTR variants, NGS has limitations that relate to interpretation of the clinical significance of detected mutations, many of which can be expected to be novel in SA population. The optimal and most cost-effective approach and strategy to molecular confirmation of CF in SA is under investigation. Further research employing NGS is needed to define the complete spectrum of CFTR variants in the SA population.
Sweat test reference ranges
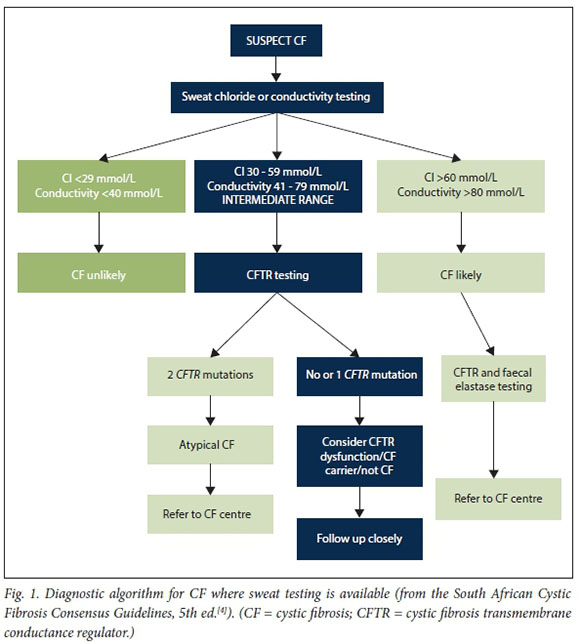
A sweat chloride concentration >60 mmol/L is the hallmark of typical or classic CF. Abnormal CFTR function in sweat glands manifests as impaired salt (NaCl) reabsorption and is the physiological basis of the sweat test. The recognition of milder or atypical forms of CF associated with mild CFTR mutations and residual CFTR function has led to revision by the International Cystic Fibrosis Foundation of the lower limit of the sweat chloride reference range in the intermediate zone to 30 mmol/L for all ages (previously 40 mmol/L) (Table 1).[5]
Sweat conductivity is another diagnostic test commonly performed in SA and in some circumstances the only type of 'sweat test' available. It is important that clinicians understand the difference between sweat chloride concentration and sweat conductivity. Sweat conductivity measures the NaCl equivalent concentration of all ionised molecules in sweat, not only chloride. Sweat conductivity is therefore on average 15-20 mmol/L higher than the equivalent sweat chloride concentration. Sweat conductivity is widely accepted as a good screening test for CF and levels of >50 mmol/L should be followed up with measurement of sweat chloride. In most cases of typical CF, sweat conductivity levels are conclusively positive. However, in mild or atypical CF, clear reference ranges for conductivity levels are lacking and may lead to misdiagnosis of CF. The MSAC therefore strongly recommends that all laboratory services in SA measure and report only sweat chloride, and use conductivity just as a screening tool. In circumstances where sweat testing is not available, the MSAC recommends measuring faecal elastase as a screening test for pancreatic-insufficient (PI) CF, which occurs in 80 - 85% of all CF cases. A normal faecal elastase level therefore does not exclude pancreatic-sufficient CF. Where PI CF is confirmed or there is strong clinical suspicion of CF collecting a blood sample for DNA testing is recommended. As faecal elastase or genetic results may take several weeks to return, we strongly advise clinicians to empirically commence pancreatic enzyme replacement therapy and other CF-related interventions while waiting for these results if the clinical suspicion of CF is high. Diagnostic algorithms for the diagnosis of CF in SA are shown in Figs 1 and 2. Furthermore, appropriate sibling and family screening through genetic counselling services is recommended in all people newly diagnosed with CF.

Novel CF treatments
Advances in the molecular biology of the CFTR molecule have identified six distinct classes of CFTR mutations, each associated with different causes and severity of CFTR dysfunction and phenotypic expression.[7] This has led to the development of breakthrough novel therapies that target the underlying CFTR defect at multiple levels, placing CF at the forefront of precision medicine.[7,8] Clinical trials of the CFTR modulator drugs ivacaftor, which targets the G551D mutation, and lumicafor/tezacaftor, which targets the p.Phe508del mutation, have reported mild to moderate beneficial effects in outcomes including pulmonary function, pulmonary exacerbations and nutrition.[9-11]The implications for identifying which CFTR mutations each person with CF carries now go beyond simply diagnosis and prognosis and include therapy.
Cautious optimism must be exercised with these new drugs, however. Ivacaftor, indicated for people who carry the G551D mutation (<5% of all people with CF), has shown the best clinical results. Recently published early phase 2 clinical trial results of two new-generation, small-molecule CFTR correctors, VX-445 and VX-659 in triple combination with tezacaftor and ivacaftor ('triple therapy'), show promising results in people with two copies of p.Phe508del.[12,13]None of these drugs are currently registered and licensed in SA, and until the cost (currently USD300 000 per annum) is substantially reduced, it is unlikely that medical schemes or the public health sector will fund these drugs in the near future. MSAC and the CF community are engaging with international pharma to advocate for affordable access to these new agents in SA.
The South African Cystic Fibrosis Registry Initiative (SACFRI).
In line with international practice, MSAC in partnership with the SAC FA launched the SACFRI in April 2018. This initiative aims to document and monitor data relating to the diagnosis and outcomes of all people diagnosed with CF in SA. There is currently little knowledge on the number of individuals with CF in SA, or on their epidemiology or outcomes. Registry data are important and useful for many reasons, including:
-
Monitoring overall health outcomes and trends in CF care in order to detect deficits or areas of care that need intervention. By comparing our data with international trends, we can identify gaps in CF care that need attention.
-
Keeping accurate statistics in SA will help us advocate for CF care to government and medical aids.
-
Planning and conducting research that will benefit the CF community, including identifying individuals who may be eligible for clinical trials of new treatments.
We encourage medical practitioners and members of the CF community to inquire about SACFRI by contacting SAC FA or their CF clinic or doctor. Appropriate precautions have been taken to ensure patient confidentiality and protection of medical and personal information, and approval has been obtained from local research ethics committees. Obtaining informed signed consent and assent from people with CF is a requirement for inclusion in the SACFRI database.
Although CF is still considered an incurable disorder in 2019, significant advances in its diagnosis, care and treatment mean that CF, previously considered universally fatal in childhood, is now a chronic condition that must be managed optimally. People with CF should look forward to leading normal productive lives, provided early diagnosis and access to quality healthcare are in place. Healthcare workers, government and health funders have an obligation to ensure that people with CF in SA are not left behind by the advancements in CF care we are witnessing in the rest of the world.
Declaration. None.
Acknowledgements. MSAC would like to thank all past and current MSAC members for the valuable contributions to advancing CF care in South Africa.
Author contributions. MZ conceptualised and wrote the manuscript. MSAC members (see Appendix 1) contributed to the content of the 5th revision of the South African Cystic Fibrosis Consensus Guidelines, published in 2017, and revised and approved the final version of this manuscript.
Funding. MSAC and SACFA have received unconditional grants from Abbott and Novartis SA to support MSAC and CF registry activities and operational expenses.
Conflicts of interest. None.
References
1. Cystic Fibrosis Foundation. Patient Registry. Annual Data Report. 2016. https://www.cff.org/Research/Researcher-Resources/Patient-Re gistry/2016-Patient-Registry-Annual-Data-Report.pdf (accessed 19 November 2018). [ Links ]
2. Goldman A, Graf C, Ramsay M, Leisegang F, Westwood AT. Molecular diagnosis of cystic fibrosis in South African populations. S Afr Med J 2003;93(7).518-519. [ Links ]
3. Westwood A. The prognosis of cysticfibrosis in the Western Cape Province of South Africa. A 33 year study. Doctor of Medicine thesis. Cape Town. University of Cape Town, 2005rchapter 9. [ Links ]
4. Zampoli M, Morrow B. The South African Cystic Fibrosis Consensus Guidelines. 5th ed. 2017. http://www.sacfa.org.za/wp-content/uploads/2017_09_14_CF_Consensus_Guidelines_2017.pdf (accessed 19 November 2018). [ Links ]
5. Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis. Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr 2017,181(Suppl).-S4-S15.el. https://doi.org/10.1016/j.jpeds.2016.09.064 [ Links ]
6. Stewart C, Pepper MS. Cystic fibrosis in the African diaspora. Ann Am Thorac Soc 2017,14(1).1-7. https://doi.org/10.1513/AnnalsATS.201606-481FR [ Links ]
7. Quon BS, Rowe SM. New and emerging targeted therapies for cystic fibrosis. BMJ 201f>,352:i859. https://doi.org/10.1136/bmj.i859 [ Links ]
8. De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med 2016:4(8):662-674. https://doi.org/10.1016/S2213-2600(16)00023-0 [ Links ]
9. Elborn JS, Ramsey BW, Boyle MP, et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup. A pooled analysis. Lancet Respir Med 2016,4(8).617-626. https://doi.org/10.1016/S2213-2600(16)30121-7 [ Links ]
10. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551Dmutation.N Engl J Med2011065(18)(1663-1672. https://doi.org/10.1056/NEJMoall05185 [ Links ]
11. Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med 2017,377(21).2013-2023. https://doi.org/10.1056/NEJMoal709846 [ Links ]
12. Keating D, Marigowda G, Burr L, et al. X-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 2018,379.1612-1620. https://doi.org/10.1056/NEJMoal807120 [ Links ]
13. Davies JC, Moskowitz SM, Brown C, et al. VX-659-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 2018-,379:1599-1611. https://doi.org/10.1056/NEJMoal807119 [ Links ]
 Correspondence:
Correspondence:
M Zampoli
m.zampoli@uct.ac.za
Accepted 13 September 2018
Members of the South African Cystic Fibrosis Medical and Scientific Advisory Committee (MSAC)
Prof. Brenda Morrow, Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town; Prof. Refiloe Masekela, Department of Paediatrics and Child Health, College of Health Sciences, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban; Dr Greg Calligaro, Division of Pulmonology, Department of Medicine, Groóte Schuur Hospital and Faculty of Health Sciences, University of Cape Town; Dr Cathy Baird, Charlotte Maxeke Academic Hospital and Milpark Netcare Private Hospital, Johannesburg; Dr Susan Klugman, Department of Paediatrics and Child Health, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg; Prof. Mervyn Mer, Division of Pulmonology and Critical Care Medicine, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg; Dr Paul Gebers, private practice, Port Elizabeth; Dr Carla Els, private practice, Johannesburg; Dr Dave Richards, private practice, Johannesburg; Dr Jonathan Egner, private practice, Durban; Sarah Walters, genetic counsellor, Ampath Laboratories; Dr Julie Morrison, Department of Paediatrics and Child Health, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town; Dr Alta Terblanche, Department of Paediatrics and Child Health, School of Medicine, Faculty of Health Sciences, University of Pretoria; Mr Alain Wolf, South African Cystic Fibrosis Association (SACFA); Mr Alan Dunn, SACFA.


{kind=link}