










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.108 n.9 Pretoria Sep. 2018
http://dx.doi.org/10.7196/samj.2018.v108i9.13335
IN PRACTICE
CASE REPORT
Complete resolution of apparently definite radiological and histological usual interstitial pneumonia
D SimonI; E M IrusenII; B W AllwoodIII; J RigbyV; C F N KoegelenbergIV
IMB ChB, FCP (SA), MMed (Int), CertPulm (SA) Phys, MPhil (Pulmonology); Division of Pulmonology, Department of Medicine, Faculty of Medicine and Health Sciences, Stellenbosch University and Tygerberg Academic Hospital, Cape Town, South Africa
IIMB ChB, FCP (SA), FCCP, PhD; Division of Pulmonology, Department of Medicine, Faculty of Medicine and Health Sciences, Stellenbosch University and Tygerberg Academic Hospital, Cape Town, South Africa
IIIMB ChB, FCP (SA), CertPulm (SA), PhD; Division of Pulmonology, Department of Medicine, Faculty of Medicine and Health Sciences, Stellenbosch University and Tygerberg Academic Hospital, Cape Town, South Africa
IVMB ChB, FCP (SA), MMed (Int), CertPulm (SA), FRCP (UK), PhD; Division of Pulmonology, Department of Medicine, Faculty of Medicine and Health Sciences, Stellenbosch University and Tygerberg Academic Hospital, Cape Town, South Africa
VMB BCh, FCPath (Anat) (SA); Division of Anatomical Pathology, Department of Pathology and National Health Laboratory Service, Faculty of Medicine and Health Sciences, Stellenbosch University and Tygerberg Academic Hospital, Cape Town, South Africa
ABSTRACT
Idiopathic pulmonary fibrosis is considered to be the most common form of pulmonary fibrosis. It is a progressive and irreversible disease with a reported median survival of ~3 years. The pathological correlate is usual interstitial pneumonia (UIP), and although antifibrotic agents can slow down lung function decline, they do not completely reverse the disease process. To date, there have been no case reports describing reversal of UIP. We present a case where both the imaging and histology were compatible with definite UIP, yet it reversed with immunosuppressive therapy without the use of antifibrotic agents.
Idiopathic pulmonary fibrosis (IPF) is considered to be the most common form of pulmonary fibrosis.[1] It is progressive and irreversible, with a reported median survival of ~3 years.[1,2] The pathological correlate is usual interstitial pneumonia (UIP). The presentation tends to be insidious, with imaging features that are irreversible and progressive.[2,3] Antifibrotic agents are currently the only medication shown to slow lung function decline; however, they do not reverse the disease process.[4,5] To date, there have been no case reports describing reversal of UIP. We present a case where both the imaging and histology were compatible with definite UIP, yet it reversed with immunosuppressive therapy without the use of antifibrotic agents.
Case report
A 59-year-old man presented with a 2-week history of worsening dyspnoea, dry cough and associated fever. He was a non-smoker, healthy prior to this presentation, and maintained a very active lifestyle, including participating in an adventure cave run 5 months before the onset of his current symptoms. Before referral, he was treated unsuccessfully with antibiotics.
Clinical examination revealed a cyanotic patient with a respiratory rate of 24 breaths per minute and an oxygen saturation of 66% on room air. He was afebrile and had a normal blood pressure and pulse rate. He had bibasal crackles, had no finger clubbing, was not wasted and had no generalised lymphadenopathy. The rest of the physical examination was unremarkable, with no features of collagen vascular diseases. His initial partial pressure of arterial oxygen (PaO2) was 7 kPa, with a partial pressure of carbon dioxide in arterial blood (PaCO2) of 3.4 kPa on room air, necessitating non-invasive positive pressure ventilation.
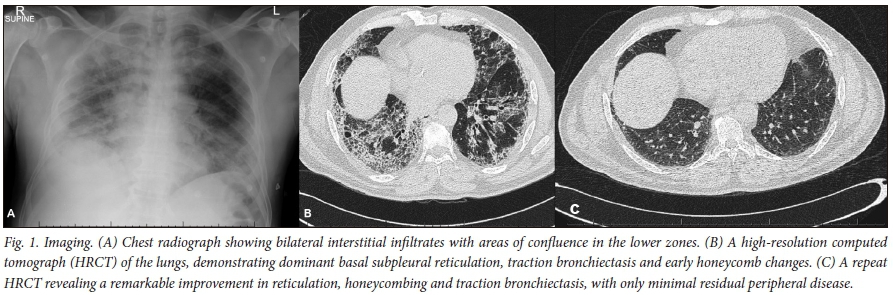
His initial chest radiograph demonstrated bilateral diffuse reticular shadowing (Fig. 1A). He tested negative for HIV and had a C-reactive protein of 147 mg/L (normal <10 mg/L). Sputum microscopy and culture revealed no bacteria or fungi, and sputum GeneXpert (Cepheid, USA) was negative. His urinary antibody test for Legionella pneumonia and all serological autoimmune tests were negative.
On presentation, he was treated empirically with intravenous antibiotics and antifungals, with no clinical response.
His initial pulmonary function testing was suggestive of a restrictive defect, with a ratio of volume of gas exhaled during the first second of forced expiration (FEV1) to the maximum volume of gas exhaled from the position of maximal inspiration by means of maximally forced vital capacity (FVC) of 0.9. His total lung capacity (TLC) was 48.8% predicted, and diffusion capacity for carbon monoxide (DLCO) was severely impaired at 28.2%.
A high-resolution computerised tomogram (HRCT) revealed peripheral reticulation, extensive subpleural honeycombing, and traction bronchiectasis predominating in the lower lobes (Fig. 1B).
Because of the subacute presentation and lack of obvious exposure history we opted to perform an open lung biopsy. Histology revealed interstitial expansion with fibroblastic foci and architectural distortion with mild to moderate inflammation, as well as uninvolved foci with alveolar dilatation (Fig. 2). There was no granulomatous inflammation, multinucleated giant cells, foreign material or eosinophilic infiltrate. All the bacteriological, viral and fungal cultures of the biopsied tissue were negative, and the histological diagnosis of definite UIP was made.[2]

The patient was provisionally diagnosed with IPF, given the absence of a secondary cause of UIP. However, there was a perplexing discordance between the degree of fibrosis and the reported duration of symptoms. We elected for an in-hospital trial of prednisone 60 mg, which was weaned during his 6-week admission because of a good clinical response. Azathioprine 150 mg was started on discharge.
After 3 months, his dyspnoea had completely resolved and he had begun cycling. Objectively, his FVC had improved to 83%, his TLC to 85% and his DLCO to 81.3%. His lung function essentially normalised at 1-year follow-up. A repeat HRCT (Fig. 1C) revealed a remarkable improvement in reticulation, honeycombing and traction bronchiectasis, with only minimal residual peripheral disease remaining.
All immunosuppressive therapy was discontinued after his 6-month review, and he remained asymptomatic. To date, he has not developed an autoimmune disease, and a repeat autoimmune serological screen performed 6 months after discontinuing therapy remained negative.
Discussion
In contrast to our patient's subacute clinical presentation, IPF is classically associated with a more insidious onset and chronic course.[1] Our patient's convincing radiological and histopathological diagnosis of IPF was discordant with his clinical presentation, causing us to consider alternative diagnoses. His history of cave running 5 months before, although tantalising for a fungal infection, was deemed too long before presentation to be the cause in an immunocompetent individual, and was additionally excluded on both histology and culture. It is possible that such an exposure could initiate a subacute hypersensitivity pneumonitis; however, the lack of any confirmatory histology or suggestive radiology made this entirely speculative.
Similarly, an evolving autoimmune disease seemed highly unlikely owing to the complete lack of clinical or serological evidence up to 18 months after presentation. Other secondary causes for the UIP pattern were diligently excluded at presentation.
The most remarkable feature of this case was the almost complete resolution of what was labelled as fibrosis both on imaging and histology. Acute exacerbations of a chronic interstitial lung disease can respond to immunosuppression; however, normalisation of pulmonary function tests and chronic radiological abnormalities would be extremely unusual. Undoubtedly, subacute hypersensitivity pneumonitis (HP) can resolve completely with immunosuppression. Chronic HP can cause a UIP-type pattern and mimic IPF, but the extent of fibrosis in such patients is significant, and residual fibrosis and symptoms after treatment are usual. Therefore, both acute and chronic HP diagnoses are problematic.
Despite fulfilling the diagnostic criteria for definite, biopsy-proven IPF, idiopathic nonspecific interstitial pneumonia or HP is the most plausible alternative explanation for our patient's presentation. However, using either of these diagnostic labels is equally flawed.
This case highlights that a degree of clinical acumen is still needed when following the guidelines for chronic lung diseases. An appreciation of the deviation of our patient's presentation from the usual natural history of IPF prompted a trial of immunosuppressive therapy, which is not currently recommended, and has been considered harmful in IPF.[6] The resolution of proven fibrosis remains perplexing, but his clinical improvement cannot be debated.
Teaching points
-
Clinical acumen is paramount when following guidelines related to diffuse parenchymal lung diseases.
-
IPF is a diagnosis of exclusion.
-
There are many causes of radiological UIP, e.g. IPF, chronic HP and rheumatoid lung disease.
Acknowledgements. We acknowledge the Division of Cardiothoracic Surgery, Department of Surgical Sciences, Tygerberg Hospital and Stellenbosch University, where the open lung biopsy was performed.
Author contributions. DS, BWA and CFNK wrote the manuscript, which was reviewed by all co-authors.
Funding. None.
Conflicts of interest. None.
References
1. Koegelenberg CFN, Ainslie GM, Dheda K, et al. Recommendations for the management of idiopathic pulmonary fibrosis in South Africa: A position statement of the South African Thoracic Society. J Thor Dis 2016;8(12):3711-3719. https://doi.org/10.21037/jtd.2016.12.05 [ Links ]
2. Raghu G, Collard HR, Egan JJ, Martinez FJ; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183(6):788-824. https://doi.org/10.1164/rccm.2009-040GL [ Links ]
3. Collins CD, Wells AU, Hansell DM, et al. Observer variation in pattern type and extent of disease in fibrosing alveolitis on thin section computed tomography and chest radiography. Clin Radiol 1994;49(4):236-240. [ Links ]
4. Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2005;171(9):1040-1047. https://doi.org/10.1164/rccm.200404-571OC [ Links ]
5. Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2010;35(4):821-829. https://doi.org/10.1183/09031936.00005209 [ Links ]
6. Raghu G, Anstrom KJ, King TE jun., Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetyl-cysteine for pulmonary fibrosis. N Engl J Med 2012;366(21):1968-1977. https://doi.org/10.1056/NEJMoa1113354 [ Links ]
 Correspondence:
Correspondence:
D Simon
siya_bonga@icloud.com
Accepted 17 May 2018


{kind=link}