










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.108 n.8 suppl.1 Pretoria Aug. 2018
http://dx.doi.org/10.7196/samj.2018.v108i8.13500
RESEARCH
Molecular and cellular oncogenic mechanisms in hepatocellular carcinoma
M SetshediI, II; M AnderssonIII, IV; M M KgatleV, VI; L R RobertsVII
IMB ChB, FCP, MPH, Cert Gastroenterology, PhD;Department of Medicine, University of Cape Town, Observatory, South Africa
IIMB ChB, FCP, MPH, Cert Gastroenterology, PhD; MRC HIU, Weatherall Institute of Molecular Medicine, University of Oxford, United Kingdom
IIIMBBS, DTM&H, MRCP, FRCPath, MD; Division of Medical Virology, Department of Pathology, Stellenbosch University, Tygerberg, Cape Town, South Africa
IVMBBS, DTM&H, MRCP, FRCPath, MD; Oxford University Hospitals NHS Foundation Trust, Medical Microbiology and Virology, Oxford, United Kingdom
VBSc, BSc (Med) Hons, MSc (Med), PhD; Department of Medicine, University of Cape Town, Observatory, South Africa
VIBSc, BSc (Med) Hons, MSc (Med), PhD; Department of Biochemistry, University of Oxford, United Kingdom
VIIMB ChB, PhD; Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, Rochester, MN, USA
ABSTRACT
Hepatocellular carcinoma (HCC), as the fifth most diagnosed cancer in the world and the third leading cause of death, is a global health concern. Research stimulated by the dismal prognosis of HCC has led to significant advances in the understanding of its aetio-pathogenesis. Dysregulation of genetic, epigenetic and signalling pathways as well as tumour immunological escape mechanisms are implicated in the development of HCC. This review summarises the current knowledge of these mechanisms and argues that it is only through further understanding of their role in hepatocarcinogenesis, that new effective therapies can be developed.
Hepatocellular cancer (HCC) is the most common primary cancer of the liver and was accountable for 782 000 incident cases in 2012. Of these cases, an alarming 745 000 patients died.1 These figures attest to the dismal post-diagnosis outlook (3.4 months median survival) with or without screening.2 The chief risk factor for HCC in most patients is cirrhosis. In 80 - 90% of all cases, HCC is due to hepatitis B (HBV) and C (HCV) virus infections. Furthermore, because of diabetes and obesity, there is an increasing prevalence of non-alcoholic fatty liver disease (NAFLD), which is now the most common liver disorder in North America.3 The annual incidence of NAFLD-related HCC has increased by 9% per year from 2004 to 2009.4 Notable co-factors, such as alcohol, primary or secondary iron overload and aflatoxin contamination of stored food products are thought to play a synergistic role in promoting hepatocarcinogenesis, particularly in the context of HBV and non-alcoholic steatohepatitis.5,6 HCC is a complex disease due to its heterogeneity: from a clinical perspective in addition to the multiple aetiological risk factors, HCC typically has a prolonged asymptomatic phase early in the disease, and thus tends to present late with an aggressive phenotype that may not be amenable to currently available therapies. In terms of diagnosis and response to treatment, HCC displays several histopathological phenotypes, including, but not limited to, well and poorly differentiated tumours, and tumours exhibiting features of both hepatocellular and cholangiocarcinoma.7 At the molecular level, HCC is characterised by dysregulation of multiple genetic, epigenetic and signalling pathways that interact with the tumour microenvironment to facilitate tumour initiation, progression and metastasis. This review aims to concisely elucidate the current understanding of the molecular and cellular pathogenesis of HCC outlined in Table 1.
Cancer phenotype
Normal cells are originally embryonically and developmentally equivalent. However, they undergo a sequential process of cell fate determination, proliferation and differentiation. This process is dependent on extra- and intracellular interactions that are governed by various signalling pathways.8 Physiologically, these pathways are activated during early life but are dormant in adulthood. However, following inflammation or another insult, these pathways are re-activated, resulting in dysregulated cellular signalling, which accounts for the metamorphosis from normal to transformed malignant cells. Cancer cells, therefore, owing to genomic instability and/or mutations induced by cellular damage, have a distinct phenotype. These cells acquire the ability to: (i) autonomously proliferate (i.e. they are independent of external mitogenic signals); (ii) avoid both anti-growth and apoptotic signals, giving them a growth advantage; and (iii) deregulate certain cellular functions responsible for cellular growth and differentiation.9 Furthermore, cancer cells exploit signalling pathways to penetrate surrounding healthy tissue including the vascular epithelium, resulting in metastases to distant sites. Moreover, there is ample evidence that by their existence, cancer cells are able to suppress T-cell cytotoxicity and related immune mechanisms. This aggressive phenotype underpins the hallmark of carcinogenesis and explains malignant transformation. Therapies aimed at halting this autonomy need to be able to keep up with the many mechanisms involved; therefore, an understanding of the pathways is a pre-requisite.
Current concepts of hepatocarcinogenesis
Multi-step process
The currently accepted model of hepatocarcinogenesis is a multistep process from tumour initiation to established malignancy. The evidence for step-wise progression of HCC is that normal hepatocytes are transformed to pre-neoplastic lesions, which occur in the form of dysplastic foci and nodules (DN) (<1 mm and >1 mm, respectively).10 With ongoing chronic inflammation, these early lesions progress to low- and high-grade dysplasia, both of which have the potential to progress to HCC (Fig. 1).11,12 The underlying mechanism of sequential progression is incompletely understood but is thought to be due to progressive hepatocyte dedifferentiation due to impaired liver-specific gene expression and the alteration of numerous signalling pathways, leading to dysregulated cell proliferation and resistance to apoptosis.13 In patients with HCC gene expression, patterns of cell proliferation markers and anti-apoptotic genes were significantly higher in the group of patients with poorer prognosis, lending credence to their significance in HCC pathogenesis.14,15
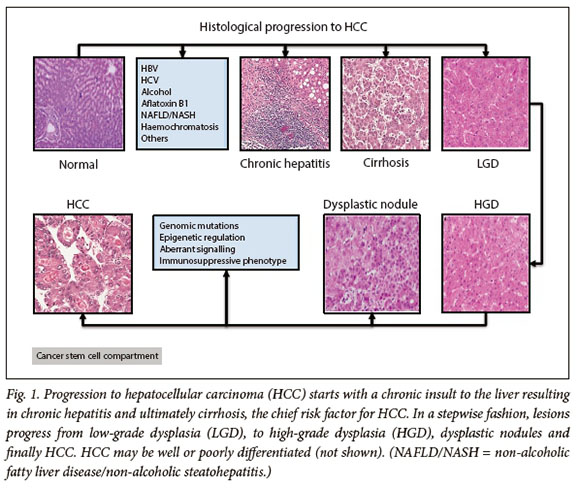
Cancer stem cells
The long-held stochastic model of HCC pathogenesis states that damaged cells in tissue can randomly result in tumour initiation and/or growth. An attractive alternative theory is that within a tumour, a small population (<1%) of cells have phenotypic characteristics of adult progenitor stem cells, in that they have an inordinate capacity to autonomously proliferate and self-renew. As a result of a loss of regulation these cells accumulate, forming the bulk of the tumour (also called the cancer stem cell compartment) and are implicated in tumour initiation and maintenance.16,17 The expression of liver stem cell markers has been found in large numbers of human HCC, suggesting that human stem cells give rise to HCC.18 In fact CD133, one of the tumorigenic stem cell markers, was found in both HCC cell lines and primary tissues.19,20 Furthermore, the clinical relevance of these stem cells is that they have enhanced chemotherapy and radiotherapy resistance and are therefore typically associated with metastases and relapse.20,21 In order to more effectively attain better survival outcomes from currently available therapies including immunotherapy, further work into understanding the genetic and signalling pathways that regulate this cellular compartment is urgently required.
Molecular pathways involved
The requirement for carcinogenesis is a permissive milieu where genes and signalling pathways that regulate the fate of all cells, i.e. differentiation, proliferation and death ,are altered. In this context, mutations of oncogenes or tumour suppressor genes in HCC become more important determinants (Fig. 2).
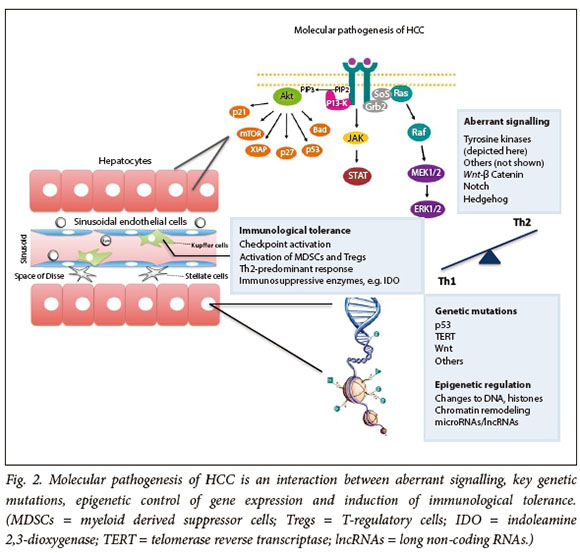
Genetic factors
Mutations of the telomerase promoter
Telomeres are protective nucleotide sequences capping the ends of chromosomes. These are particularly significant in the context of the liver in that the reparative capability of the telomerase enzyme affords hepatocytes their near-inexhaustible regenerative ability. However, when chronic inflammation occurs, the rate of telomere shortening is accelerated, which co-operates with inactivating mutations of telomerase to contribute to the development of cirrhosis.22,23 Under normal circumstances the telomerase enzyme is switched off to rid the body of senescent or abnormal cells. In HCC, however, mutations of the promoter region of the telomerase reverse transcriptase (TERT) allow malignant cells to evade apoptosis, resulting in an immortal phenotype. In HCC these are the most commonly described mutations, occurring in 29 - 60% of HCCs.24,25
Mutations of the total protein 53 (TP53) pathway
TP53 has many anticancer functions including DNA repair, inhibition of G1/S cell cycle progression, and initiation of apoptosis by regulating the transcription of protective antioxidant genes and transactivating pro-oxidant genes.26 Inactivating mutations of TP53, are common in many cancers, not least in HCC, where they are present in 18 - 50% of cases.26,27 There are several variants of TP53 mutations in different cancers,28 which suggests a role for environmental influences on cancer phenotype. In HCC the most well-described TP53 mutation is a result of a transversion of G:C to T:A at codon 249, as a result of the synergism between aflatoxin B and HBV (particularly in endemic areas).27,29,30 The detection of TP53 mutant DNA in plasma is a biomarker of both AFB(1) exposure and HCC risk.
Other mutations
There are many other genes involved in HCC that regulate proto-oncogene, tumour suppressor, signaling pathway, DNA-binding and other functions; these have been reviewed elsewhere.7,31
Epigenetic factors
Epigenetics refers to heritable alterations in gene expression not due to changes of the genome itself that, under normal conditions, are used by the body to control processes such as X chromosome inactivation.32,33 Evidence exists, however, to suggest that changes in the epigenome are associated with HCC initiation and progression.34 Epigenetic control is conferred by several mechanisms.
Modifications to DNA
The generally accepted mechanisms by which carcinogenesis occurs are global hypomethylation resulting in activating mutations of oncogenes, e.g. in the Wnt pathway.35 In HCC, however, the hypermethylation of promoter regions of tumour suppressor genes is more typical and results in their silencing36 by either inhibiting the interaction of transcription factors with their promoter, or binding of methyl-CpG binding domain proteins, to methylated DNA.37,38 Genome-wide methylation profiling studies have identified multiple hypermethylated gene promoters including adenosis polyposis coli (APC) and others in HCC tumours compared with surrounding non-tumour tissue.39-44 This is clinically relevant because, for instance, low levels of sphingomyelin phosphodiesterase 3 (SMPD3), a potent tumour suppressor, were three times more likely to be associated with early recurrence of HCC after curative surgery in an independent patient cohort.45,46 In this context, therefore, methylation profiling holds promise in terms of predicting patients who are more likely to progress to HCC.
Modifications to histones
Post-translational modifications resulting in an open or closed configuration of histone proteins, which affects their accessibility, have a significant effect on the 'on' or 'off' state of gene expression.47 While acetylation by histone acetyltransferases (HATs) causes activating transcription of genes,48 histone deacetylases (HDACs) result in tight coiling of DNA around the histones, leading to transcriptional repression.49 By contrast, methylation confers a dual role of activation or repression, which is context-specific. For example, tri-methylation of lysine 4 (K4) and 36 on histone 3 (H3K4me3 and H3K36Me3) are transcriptionally active start sites (TSS) of active genes.50-52 Histone H3 lysine 4 (H3K27me3) is significantly elevated in patients with HCC, and this correlates with a poor prognosis (3.5-fold increased risk of death) as a result of aggressive tumour features, including vascular invasion, large tumour size and poor differentiation.53,54
Chromatin remodelling
Epigenetic gene silencing can also be mediated by a group of chromatin-modifying proteins known as polycomb repressive complexes (PRCs).
PRC1 and 2 are the chief epigenetic repressors involved in the maintenance of stem and adult cells and regulate repression by ubiquitination of group histone 2A lysine 119 (H2AK119), and tri-methylation of histone H3 lysine 27 (H3K27), respectively.55 Increased levels of EZH2, one of the components of the PRC2 complex, correlate with an aggressive HCC phenotype, associated with metastases and poor prognosis.54,56 Mechanistically, EZH2 silences Wnt antagonists, thereby activating Wnt -β catenin signalling to promote cancer progression,57 whereas knockdown causes re-expression of tumour suppressor mRNAs,58 paying credence to its biological relevance.
Regulation by micro- and long non-coding RNAs
MicroRNAs (miRNAs) are 17 - 25-nucleotide-long non-coding RNA molecules that up- or de-regulate post-transcriptional gene expression by modifying the stability of or degrading mRNA.59,60 miRNAs/miR are important in the context of carcinogenesis because they regulate differentiation, development and oncogenesis.61 In addition to regulating various cellular processes, miRNAs are epigenetic modulators by targeting mRNAs of epigenetic regulators including DNA methyltransferase 3 alpha (DNMT3A), DNA methyltransferase 3 Beta (DNMT3B), polycomb mRNAs, EZH2 (as shown above), BMI1 and HDAC4.62,63 miR-122 is most abundant in the liver and is frequently downregulated in HCC, which suggests its role as a tumour suppressor.64,65 Additional miRNAs that function as tumour suppressors include miR-26a, miR-26b, miR-125b, miR-140-5p, miR-217, miR-138, miR-148b, miR-325, miR-451. 36,63 These are decreased in HCC, and are associated with a poor prognosis, therefore they may function as potential biomarkers for HCC. Reduced miR-26 expression correlates with shorter survival, but encouragingly, these patients are more likely to respond to interferon alpha therapy, making it an ideal candidate for predicting response to therapy.66
Another group of non-coding RNAs (about 200 nucleotides in length) that regulate gene expression are the long non-coding RNAs (lncRNAs). Twenty percent of lncRNAs are associated with PRC2, through which they recruit and guide chromatin-modifying complexes to specific genomic regions to regulate gene expression.67 Other mechanisms of gene regulation by non-coding RNAs involve downregulation of tumour suppressor gene, activation of cell cycle function and chromatin reprogramming to promote metastases.68 These novel epigenetic regulators offer exciting opportunities for new therapies for HCC.
Signalling pathways
Several signalling pathways involved in all aspects of cell fate determination are exploited by cancerous cells to favour proliferation, growth, invasiveness and metastases. Although for clarity these will be discussed in separate sections based on their effect in tumour promotion, it is important to note that there is crosstalk between these pathways to mediate their effects. For example, Chung et al.69show evidence of tripartite signal induction of the insulin/MAPK/ ERK, Wnt and Notch pathways in a double transgenic mouse model of HBV/HBx protein to result in hepatocarcinogenesis.
Receptor tyrosine kinase pathways Pathways involved with growth
The tyrosine kinases are key regulators of cellular proliferation, differentiation, survival, metabolism, migration and cell cycle control.70,71 Binding of insulin-like growth factors (IGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF/c-MET), transforming growth factor (TGF), basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), and vascular endothelium growth factor (VEGF) to their corresponding receptors initiates and activates signalling cascades that promote growth and differentiation. In the context of liver regeneration following an insult, these pathways are upregulated, resulting in aberrant signaling affecting multiple pathways,72 promoting cancer initiation and progression. Downstream, the intracellular mediators of these pathways are the Ras-mitogen-activated protein kinase (MAPK) or extracellular signalling regulated kinase (ERK), phosphatidylinositol 3-kinase (PI3K)/Akt kinase signalling pathways and JAK/STAT pathways that induce transcription of cell-proliferating genes via proto-oncogene cFos and transcription factor activator protein (AP-1).73 Both IGF-I and IGF-II (increased expression in 12 - 44% of HCC) acting through the IGF-1 receptor (IGF-1R) are involved in the development and progression of HCC.74 Similarly, EGFR, HGF and c-Met (a transmembrane tyrosine kinase) are implicated in aggressive HCC, associated with a poor prognosis.75,76
Pathways involved with angiogenesis
HCC is a highly vascular tumour with high metastatic potential. This is partly due to activation of VEGF (through VEGFR2), PDGF (through FGFR-1) and bFGF signalling pathways involved in neo-vascularisation, invasion and metastases.77-79 High levels of VEGF are associated with postoperative recurrence and, therefore, poor prognosis in HCC.80,81 Notably, bFGF intersects with VEGF to synergistically activate angiogenic pathways,82 suggesting that it may indeed be a target for drug resistance against VEGF-targeted therapies. Furthermore, high preoperative serum bFGF levels are predictive of invasive tumour and early postoperative recurrence in patients undergoing resection, making this a potentially useful clinical biomarker.79 These pathways can be inhibited by sorafenib; it is the only multi-tyrosine kinase inhibitor that targets VEGFR 1-3, PDGFR-β, c-kit, Flt3 and p38, and remains the only one approved for use in clinical practice for unresectable HCC. Sorafenib, however, confers only a 2 - 3-month survival benefit, highlighting the critical need for new therapies in this group of patients.79,83 Newer trials have been designed that target either multiple tyrosine kinase inhibitors (TKIs) simultaneously or specific TKIs such as c-MET inhibitors or TGFpR in HCC sub-populations, with promising early results.84
Pathways involved with cell differentiation Wnt-β catenin
Wnt-β catenin is one of the most studied and commonly implicated aberrant pathways in early HCC. Due to the multitude of ligands and receptors involved, it renders the effects of signalling through this pathway unpredictable, with some binding resulting in inhibition and others activation of signalling. Notwithstanding, canonical Wnt signalling results in translocation of beta-catenin into the nucleus binding with TCF/LEF transcription factors coding for genes involved in cell proliferation angiogenesis, anti-apoptosis, and the formation of extracellular matrix (ECM), causing Wnt upregulation.85,86 Mechanisms of Wnt activation include somatic mutations of CTBBB1, AXIN1 and AXIN2, as well as inactivation of tumour suppressor adenosis polyposis coli (APC), which mimic pathway activation. Other mechanisms include epigenetic control of proteins of Wnt signalling.
Notch
The Notch pathway is a primitive and highly conserved pathway that is crucial in mammalian embryogenesis, cell fate determination, liver repair and regeneration.87 Its role in hepatic carcinogenesis is emerging; of the four Notch receptors, Notch 4 is well characterised as the most oncogenic, whereas Notch 1 may be either up- or downregulated. The function of Notch 3 appears minimal in HCC and that of Notch 4 is related to invasiveness and metastases rather than tumour initiation.88 Similar to the Wnt pathways, aberration in the pathway results in activation or inhibition of oncogenes and tumour suppressor genes, respectively, and cross-talk with other pathways, the net effect of which may explain the heterogeneous phenotypic expression. Notch signalling is aberrantly upregulated in HCC compared withnormal liver tissues.89
Hedgehog
Activation of Hedgehog signalling was shown to be oncogenic for the first time when blocking of this pathway resulted in reduced proliferation, apoptosis and repressed C-myc and cyclin D expression, both in human HCC samples and liver cancer cell lines.90 Glioma-associated oncogene homolog-1 (Gli-1), a marker of Hedgehog pathway activation, is correlated to invasiveness and the risk of metastases in HCC. Inhibition of this pathway by small interfering RNA significantly suppressed adhesion, motility, migration and invasion of liver cancer cell lines and the expression and activities of both matrix metalloproteinases-2 and 9 (MMP-2 and MMP-9).91,92 Indeed, Hedgehog activation may be useful as a biomarker to delineate malignant from adjacent normal tissue and thus may be a useful target for local therapy,91 particularly as an inducer of apoptosis.93 Other mechanisms by which Hedgehog is oncogenic include the activation of MMP-9 through ERK.94 A key role of Hedgehog activation is that it is an inducer of radiation-induced liver fibrosis, which can be targeted with inhibitors to radiosensitise tumours prior to radiotherapy.95
Immunological tolerance
The recent discovery and therapeutic potential of checkpoint inhibitors attests to the significant role of the immune system in the pathogenesis of HCC. The liver is an immunologically rich organ, elegantly poised to deal with gut-derived pathogens from the portal vein. More importantly, however, is its adaptive ability to effect immune tolerance as a protective mechanism to avoid excessive liver injury. This is achieved through several key immunological mechanisms: liver sinusoidal endothelial cells have a high expression of programmed death ligand 1 (PDL-1) and low expression of co-stimulatory CD80 and CD86,96,97 and downregulate MHC molecules and dendritic cell activation,98,99 thus curtailing their cytotoxic ability. In HCC specifically, there is immune exhaustion, typified by enhanced expression of co-inhibitory molecules PDL-1, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte-activation gene 3 (LAG3) and T cell immunoglobulin domain and mucin domain 3 (TIM-3),100 including decreased expression of effector cytokines, which limits cytotoxic effectiveness.101 Additionally, within the HCC tumour microenvironment, the immune response is directed towards an immunosuppressive phenotype with the release of anti-inflammatory cytokines, interleukin-10 and TGF-ß.102 Other tumour evasion/escape strategies include the recruitment of immunosuppressive T-regulatory cells (T-regs) and monocyte-derived myeloid suppressor cells (MDSCs), and inhibitory indoleamine 2,3-dioxygenase (IDO), tryptophan 2,3-dioxygenase (TDO) and arginase-1 enzymes, which render immune cells deficient of tryptophan and arginine required for optimal functioning. The mechanisms by which malignant cells are able to thrive in this nutrient-deficient milieu are under investigation. However, a paper by Timosenko et al. 103 describes the ability of HeLa cells to upregulate an amino acid transporter, solute carrier family 1 member 5 (SLC1A5), which imports tryptophan, whereas co-cultured T-cells were unable to do so, thus disabling their cytotoxic functioning.
Conclusion
Despite decades of research into molecularly targeted therapies, including the recent advent of immunotherapy, the armamentarium against HCC is at best discouraging. None of these agents, including sorafenib, have translated into clinically meaningful improved patient survival. As such, HCC remains a deadly cancer. The understanding of all hepatocarcinogenic pathways is therefore critical to yield to new and effective therapies for HCC. Specifically, the exploration of epigenetic and immunological factors may more imminently result in faster progress towards alternative therapies. These efforts will require closer collaborations, not only between various medical disciplines, but also with basic/molecular biology scientists, immunologists, the pharmacological industry, and government bodies.
Acknowledgements. The authors would like to thank Professor Mike Kew for reviewing the manuscript.
Author contributions. MS wrote the first draft, incorporated the suggested corrections, and designed the table and graphics. All authors made suggestions to the text/meaning of the manuscript and contributed to the final submission.
Funding. None.
Conflicts of interest. None.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136(5):E359-386. [ Links ]
2. Khalaf N, Ying J, Mittal S, et al. Natural history of untreated hepatocellular carcinoma in a US Cohort and the role of cancer surveillance. Clin Gastroenterol Hepatol 2017;15(2):273-281. https://doi.org/10.1016/j.cgh.2016.07.033 [ Links ]
3. Younossi ZM, Stepanova M, Afendy M, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol 2011;9(6):524-530. https://doi.org/10.1016/j.cgh.2011.03.020 [ Links ]
4. Younossi ZM, Otgonsuren M, Henry L, et al. Association ofnonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015;62(6):1723-1730. https://doi.org/10.1002/hep.28123 [ Links ]
5. Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist 2010;15(Suppl 4):14-22. https://doi.org/10.1634/theoncologist.2010-S4-14 [ Links ]
6. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10(11):686-690. https://doi.org/10.1038/nrgastro.2013.171 [ Links ]
7. Dhanasekaran R, Bandoh S, Roberts LR. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Res 2016:F1000 Faculty Rev 879. https://doi.org/10.12688/f1000research.6946.1 [ Links ]
8. Perrimon N, Pitsouli C, Shilo BZ. Signaling mechanisms controlling cell fate and embryonic patterning. Cold Spring Harb Perspect Biol 2012;4(8):a005975. https://doi.org/10.1101/cshperspect.a005975 [ Links ]
9. Hanahan D, Weinberg RA. The hallmarks of cancer: The next generation. Cell 2000;100(1):57-70. https://doi.org/10.1016/j.cell.2011.02.013 [ Links ]
10. Kojiro M, Roskams T. Early hepatocellular carcinoma and dysplastic nodules. Semin Liver Dis 2005;25(2):133-142. https://doi.org/10.1055/s-2005-871193 [ Links ]
11. Thorgeirsson SS, Grisham JW Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002;3(4):339-346. https://doi.org/10.1038/ng0802-339 [ Links ]
12. Coleman WB. Mechanisms of human hepatocarcinogenesis. Curr Mol Med 2003;3(6):573-588. [ Links ]
13. Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene 2006;25(27):3866-3884. https://doi.org/10.1038/sj.onc.1209550 [ Links ]
14. Lee JS, Thorgeirsson SS. Genome-scale profiling of gene expression in hepatocellular carcinoma: classification, survival prediction, and identification of therapeutic targets. Gastroenterology 2004;127(5 Suppl 1):S51-55. [ Links ]
15. Lee JS, Thorgeirsson SS. Genetic profiling of human hepatocellular carcinoma. Semin Liver Dis 2005;25(2):125-132. https://doi.org/10.1055/s-2005-871192 [ Links ]
16. Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene 2004; 23(43):7274-7282. https://doi.org/10.1038/sj.onc.1207947 [ Links ]
17. Clarke MF, Dick JE, Dirks PB, et al Cancer stem cells - perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006;66(19):9339-9344. https://doi.org/10.1158/0008-5472.CAN-06-3126 [ Links ]
18. Libbrecht L, Roskams T. Hepatic progenitor cells in human liver diseases. Semin Cell Dev Biol 2002;13(6):389-396. [ Links ]
19. Yin S1, Li J, Hu C, Chen X, et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer 2007;120(7):1444-1450. https://doi.org/10.1002/ijc.22476 [ Links ]
20. Ma S, Chan KW, Hu L, et al Identification and characterization of tumorigenic liver cancer stem/ progenitor cells. Gastroenterology 2007;132(7):2542-2556. https://doi.org/10.1053/j.gastro.2007.04.025 [ Links ]
21. Piao LS, Hur W, Kim TK, et al CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett 2012;315(2):129-137. https://doi.org/10.1016/j.canlet.2011.10.012 [ Links ]
22. Calado RT, Brudno J, Mehta P, et al. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 2011;53(5):1600-1607. [ Links ]
23. Hartmann D, Srivastava U, Thaler M, et al Telomerase gene mutations are associated with cirrhosis formation. Hepatology 2011;53(5):1608-1617. https://doi.org/10.1002/hep.24217 [ Links ]
24. Nault JC, Mallet M, Pilati C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 2013;4:2218. https://doi.org/10.1038/ncomms3218 [ Links ]
25. Chen X, Chen J, Wen J, et al. Breastfeeding is not a risk factor for mother-to-child transmission of hepatitis B virus. PLoS One 2013;8(1):e55303. https://doi.org/10.1371/journal.pone.0055303 [ Links ]
26. Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene 2007;26(15):2166-2176. https://doi.org/10.1038/sj.onc.1210279 [ Links ]
27. Kew MC. Synergistic interaction between anatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int 2003;23(6):405-409. [ Links ]
28. Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991;350(6317):427-428. https://doi.org/10.1038/350427a0 [ Links ]
29. Kirk GD, Lesi OA, Mendy M, et al 249(ser) TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinoma. Oncogene 2005;24(38):5858-5867. https://doi.org/10.1038/sj.onc.1208732 [ Links ]
30. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991;350(6317):429-431. https://doi.org/10.1038/350429a0 [ Links ]
31. Bertino G, Demma S1, Ardiri A, et al. Hepatocellular carcinoma: novel molecular targets in carcinogenesis for future therapies. Biomed Res Int 2014;2014:203693. https://doi.org/10.1155/2014/203693 [ Links ]
32. Bird A. Perceptions of epigenetics. Nature 2007;447(7143):396-398. [ Links ]
33. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004;429(6990):457-463. [ Links ]
34. Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: How environmental factors influence the epigenome. Mutat Res 2011;727(3):55-61. https://doi.org/10.1016/j.mrrev.2011.04.001 [ Links ]
35. Deng YB, Nagae G, Midorikawa Y, et al Identification of genes preferentially methylated in hepatitis C virus-related hepatocellular carcinoma. Cancer Sci 2010;101(6):1501-1510. https://doi.org/10.1111/j.1349-7006.2010.01549.x [ Links ]
36. Ma L, Chua MS, Andrisani O, So S. Epigenetics in hepatocellular carcinoma: an update and future therapy perspectives. World J Gastroenterol 2014;20(2):333-345. https://doi.org/10.3748/wjg.v20.i2.333 [ Links ]
37. Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science 2001;293(5532):1068-1070. https://doi.org/10.1126/science.1063852 [ Links ]
38. Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene 2002;21(35):5496-5503. https://doi.org/10.1038/sj.onc.1205602 [ Links ]
39. Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One 2010,5(3):e9749. https://doi.org/10.1371/journal.pone.0009749 [ Links ]
40. Song MA, Tiirikainen M, Kwee S, Okimoto G, Yu H, Wong LL. Elucidating the landscape of aberrant DNA methylation in hepatocellular carcinoma. PLoS One 2013;8(2):e55761. https://doi.org/10.1371/journal.pone.0055761 [ Links ]
41. Chen HL, Lin LH, Hu FC, et al Effects of maternal screening and universal immunization to prevent mother-to-infant transmission of HBV. Gastroenterology 2012;142(4):773-781.e2. https://doi.org/10.1053/j.gastro.2011.12.035 [ Links ]
42. Narimatsu T, Tamori A, Koh N, et al. p16 promoter hypermethylation in human hepatocellular carcinoma with or without hepatitis virus infection. Intervirology 2004;47(1):26-31. https://doi.org/10.1159/000076639 [ Links ]
43. Zhang C, Li J, Huang T, et al. Meta-analysis of DNA methylation biomarkers in hepatocellular carcinoma. Oncotarget 2016;7(49):81255-81267. https://doi.org/10.18632/oncotarget.13221 [ Links ]
44. Wong IH, Lo YM, Zhang J, et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res 1999;59(1):71-73. [ Links ]
45. Nishida N, Kudo M, Nagasaka T, Ikai I, Goel A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology 2012;56(3):994-1003. https://doi.org/10.1002/hep.25706 [ Links ]
46. Revill K, Wang T, Lachenmayer A, et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology 2013;145(6):1424-1435. https://doi.org/10.1053/j.gastro.2013.08.055 [ Links ]
47. Berger SL. The complex language of chromatin regulation during transcription. Nature 2007;447(7143):407-412. https://doi.org/10.1038/nature05915 [ Links ]
48. Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev 2005;19(5):542-552. https://doi.org/10.1101/gad.1272505 [ Links ]
49. Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Ann Rev Biochem 2007;76:75-100. https://doi.org/10.1146/annurev.biochem.76.052705.162114 [ Links ]
50. Santos-Rosa H, Schneider R, Bannister AJ, et al. Active genes are tri-methylated at K4 of histone H3. Nature 2002;419(6905):407-411. https://doi.org/10.1038/nature01080 [ Links ]
51. Bernstein BE, Humphrey EL, Erlich RL, et al Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci USA 2002;99(13):8695-8700. https://doi.org/10.1073/pnas.082249499 [ Links ]
52. Bell O, Wirbelauer C, Hild M, et al Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J 2007;26(24):4974-4984. https://doi.org/10.1038/sj.emboj.7601926 [ Links ]
53. He C, Xu J, Zhang J, et al. High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum Pathol 2012;43(9):1425-1435. https://doi.org/10.1016/j.humpath.2011.11.003 [ Links ]
54. Cai MY, Hou JH, Rao HL, et al. High expression of H3K27me3 in human hepatocellular carcinomas correlates closely with vascular invasion and predicts worse prognosis in patients. Mol Med 2011;17(1-2):12-20. https://doi.org/10.2119/molmed.2010.00103 [ Links ]
55. Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 2006;6(11):846-856. https://doi.org/10.1038/nrc1991 [ Links ]
56. Sudo T, Utsunomiya T, Mimori K, et al Clinicopathological significance of EZH2 mRNA expression in patients with hepatocellular carcinoma. Br J Cancer 2005;92(9):1754-1758. https://doi.org/10.1038/sj.bjc.6602531 [ Links ]
57. Cheng AS, Lau SS, Chen Y, et al. EZH2-mediated concordant repression of Wnt antagonists promotes beta-catenin-dependent hepatocarcinogenesis. Cancer Res 2011;71(11):4028-4039. https://doi.org/10.1158/0008-5472.CAN-10-3342 [ Links ]
58. Au SL, Wong CC, Lee JM, et al. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 2012;56(2):622-631. https://doi.org/10.1002/hep.25679 [ Links ]
59. Ambros V. microRNAs: Tiny regulators with great potential. Cell 2001;107(7):823-826. [ Links ]
60. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116(2):281-297. [ Links ]
61. Schmittgen TD. Regulation of microRNA processing in development, differentiation and cancer. J Cell Mol Med 2008;12(5b):1811-1819. https://doi.org/10.1111/j.1582-4934.2008.00483.x [ Links ]
62. Wang F, Zhao YL, Ma JC, Bi SL, Zhang Y, Shen LP. Long-term efficacy of 10-12 years after being immunized with Chinese hamster ovary cell derived hepatitis B vaccine in Chinese rural communities. Vaccine 2012;30(12):2051-2053. https://doi.org/10.1016/j.vaccine.2012.01.052 [ Links ]
63. Khan FS, Ali I, AfTidi UK, Ishtiaq M, Mehmood R. Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics. Hepatol Int 2017;11(1):45-53. https://doi.org/10.1007/s12072-016-9743-4 [ Links ]
64. Kutay H, Bai S, Datta J, et al. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem 2006;99(3):671-678. https://doi.org/10.1002/jcb.20982 [ Links ]
65. Murakami Y, Yasuda T, Saigo K, et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006;25(17):2537-2545. https://doi.org/10.1038/sj.onc.1209283 [ Links ]
66. Ji J, Shi J, Budhu A, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med 2009;361(15):1437-1447. https://doi.org/10.1056/NEJMoa0901282 [ Links ]
67. Khalil AM, Guttman M, Huarte M, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA 2009;106(28):11667-11672. https://doi.org/10.1073/pnas.0904715106 [ Links ]
68. Amicone L, Citarella F, Cicchini C. Epigenetic regulation in hepatocellular carcinoma requires long noncoding RNAs. Biomed Res Int 2015;2015:473942. https://doi.org/10.1155/2015/473942 [ Links ]
69. Chung W, Kim M, de la Monte S, et al. Activation of signal transduction pathways during hepatic oncogenesis. Cancer Lett 2016;370(1):1-9. https://doi.org/10.1016/j.canlet.2015.09.016 [ Links ]
70. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature 2001;411(6835):355-365. https://doi.org/10.1038/35077225 [ Links ]
71. Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990;61(2):203-212. [ Links ]
72. Spangenberg HC, Thimme R, Blum HE. Targeted therapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2009;6(7):423-432. https://doi.org/10.1038/nrgastro.2009.86 [ Links ]
73. Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis 2007;27(1):55-76. https://doi.org/10.1055/s-2006-960171 [ Links ]
74. Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006;25(27):3787-3800. https://doi.org/10.1038/sj.onc.1209556 [ Links ]
75. Kaposi-Novak P, Lee JS, Gomez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest 2006;116(6):1582-1595. https://doi.org/10.1172/JCI27236 [ Links ]
76. Osada S, Kanematsu M, Imai H, Goshima S. Clinical significance of serum HGF and c-Met expression in tumor tissue for evaluation of properties and treatment of hepatocellular carcinoma. Hepatogastroenterology 2008;55(82-83):544-549. [ Links ]
77. Miura H, Miyazaki T, Kuroda M, et al. Increased expression of vascular endothelial growth factor in human hepatocellular carcinoma. J Hepatol 1997;27(5):854-861. [ Links ]
78. Torimura T, Sata M, Ueno T, et al. Increased expression of vascular endothelial growth factor is associated with tumor progression in hepatocellular carcinoma. Hum Pathol 1998;29(9):986-991. [ Links ]
79. Poon RT, Ng IO, Lau C, Yu WC, Fan ST, Wong J. Correlation of serum basic fibroblast growth factor levels with clinicopathologic features and postoperative recurrence in hepatocellular carcinoma. Am J Surg 2001;182(3):298-304. [ Links ]
80. Poon RT. Prognostic significance of serum vascular endothelial growth factor and endostatin in patients with hepatocellular carcinoma. Br J Surg 2004;91(10):1354-1360. [ Links ]
81. Chao Y, Li CP, Chau GY, et al. Prognostic significance of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin in patients with resectable hepatocellular carcinoma after surgery. Ann Surg Oncol 2003;10(4):355-362. [ Links ]
82. Yoshiji H, Kuriyama S, Yoshii J, et al. Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology 2002;35(4):834-842. https://doi.org/10.1053/jhep.2002.32541 [ Links ]
83. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008;48(4):1312-1327. [ Links ]
84. De Rosamel L, Blanc JF. Emerging tyrosine kinase inhibitors for the treatment of hepatocellular carcinoma. Expert Opin Emerg Drugs 2017;22(2):175-190. https://doi.org/10.1080/14728214.2017.1336538 [ Links ]
85. Buendia MA. Genetics of hepatocellular carcinoma. Semin Cancer Biol 2000;10(3):185-200. https://doi.org/10.1006/scbi.2000.0319 [ Links ]
86. Calvisi DF, Factor VM, Loi R, Thorgeirsson SS. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: relationship to phenotype and tumor grade. Cancer Res 2001;61(5):2085-2091. [ Links ]
87. Morell CM, Fiorotto R, Fabris L, Strazzabosco M. Notch signalling beyond liver development: emerging concepts in liver repair and oncogenesis. Clin Res Hepatol Gastroenterol 2013;37(5):447-454. https://doi.org/10.1016/j.clinre.2013.05.008 [ Links ]
88. Gil-Garcia B, Baladron V. The complex role of NOTCH receptors and their ligands in the development of hepatoblastoma, cholangiocarcinoma and hepatocellular carcinoma. Biol Cell 2016;108(2):29-40. https://doi.org/10.1111/boc.201500029 [ Links ]
89. Sun W, Ma L, Hao A, et al. [Predictive value of telbivudine in preventing mother-to-infant transmission of hepatitis B virus in pregnant women with high viremia]. Zhonghua Gan Zang Bing Za Zhi 2015;23(3):180-183. [ Links ]
90. Patil MA, Zhang J, Ho C, Cheung ST, Fan ST, Chen X. Hedgehog signaling in human hepatocellular carcinoma. Cancer Biol Ther 2006;5(1):111-117. [ Links ]
91. Efroni S, Meerzaman D, Schaefer CF, et al. Systems analysis utilising pathway interactions identifies sonic hedgehog pathway as a primary biomarker and oncogenic target in hepatocellular carcinoma. IET Syst Biol 2013;7(6):243-251. [ Links ]
92. Chen JS, Li HS, Huang JQ, et al Down-regulation of Gli-1 inhibits hepatocellular carcinoma cell migration and invasion. Mol Cell Biochem 2014;393(1-2):283-291. https://doi.org/10.1007/s11010-014-2071-x [ Links ]
93. Wang Y, Han C, Lu L, Magliato S, Wu T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013;58(3):995-1010. https://doi.org/10.1002/hep.26394 [ Links ]
94. Lu JT, Zhao WD, He W, Wei W. Hedgehog signaling pathway mediates invasion and metastasis of hepatocellular carcinoma via ERK pathway. Acta Pharmacol Sin 2012;33(5):691-700. https://doi.org/10.1038/aps.2012.24 [ Links ]
95. Kabarriti R, Guha C. Hedgehog signaling and radiation induced liver injury: A delicate balance. Hepatol Int 2014;8(3):316-320. https://doi.org/10.1007/s12072-014-9532-x [ Links ]
96. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol 2013;14(10):996-1006. https://doi.org/10.1038/ni.2691 [ Links ]
97. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol 2010;10(11):753-766. https://doi.org/10.1038/nri2858 [ Links ]
98. Knolle PA, Germann T, Treichel U, et al. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J Immunol 1999;162(3):1401-1407. [ Links ]
99. Schildberg FA, Hegenbarth SI, Schumak B, Scholz K, Limmer A, Knolle PA. Liver sinusoidal endothelial cells veto CD8 T cell activation by antigen-presenting dendritic cells. Eur J Immunol 2008;38(4):957-967. https://doi.org/10.1002/eji.200738060 [ Links ]
100. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12(4):252-264. https://doi.org/10.1038/nrc3239 [ Links ]
101. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis 2015;6:e1792. https://doi.org/10.1038/cddis.2015.162 [ Links ]
102. Budhu A, Forgues M, Ye QH, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature ofthe liver microenvironment. Cancer Cell 2006;10(2):99-111. https://doi.org/10.1016/j.ccr.2006.06.016 [ Links ]
103. Timosenko E, Ghadbane H, Silk JD, et al. Nutritional stress induced by tryptophan-degrading enzymes results in ATF4-dependent reprogramming of the amino acid transporter profile in tumor cells. Cancer Res 2016;6(21):6193-6204. https://doi.org/10.1158/0008-5472.CAN-15-3502 [ Links ]
 Correspondence:
Correspondence:
M Setshedi
mashiko.setshedi@uct.ac.za


{kind=link}