










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.108 n.8 suppl.1 Pretoria Aug. 2018
http://dx.doi.org/10.7196/samj.2018.v108i8.13497
RESEARCH
Current and future directions for the management of hepatitis B
G Dusheiko
FCP(SA), FRCP, FRCP (Edin); UCL Institute of Liver and Digestive Health and King's College Hospital, London, UK
ABSTRACT
Hepatitis B virus vaccination, while effective in reducing incident chronic hepatitis B in endemic regions, will not have the desired impact on the rates of end-stage liver disease in chronically infected persons. A large reservoir of chronic infection remains and needs to be managed effectively. Over three decades, interferon alpha (IFNcx), and nucleoside analogue therapies have reduced the morbidity and mortality associated with chronic hepatitis B by suppressing viral replication and retarding the progression to cirrhosis and the development of hepatocellular carcinoma (HCC). The preferential preservation of covalently closed circular (cccDNA) and capsid reverse transcriptase-cccDNA interactions during nucleoside analogue therapy currently prevent cure; the majority of patients require continuous maintenance suppressive therapy. In selected patients nucleoside analogues may be stopped. New targets for drug therapy need to be directed at inhibiting intracellular HBV replication, transcription and translation pathways to enhance the likelihood of a cure in the host. Such cures for chronic hepatitis B infection will require several synergistic therapies to achieve either complete eradication of replicative intermediates from the host (cure), or more probably, a functional cure defined as loss of hepatitis B surface antigen. Hampering such development is the lack of a proven serological surrogate for cccDNA to evaluate treatment efficacy. This review outlines the pathophysiology of the virus, the host immunological responses and current therapies. Understanding the interactions between HBV and the host remains fundamental to guide correct sequencing and combinations of treatment with either host or viral-targeting agents to achieve higher rates of cure.
HBV infection is an important worldwide cause of morbidity and mortality from cirrhosis and hepatocellular carcinoma (HCC). An estimated 257 million people are chronically infected worldwide. The disease is most prevalent in the WHO African and Western Pacific regions. The prevalence in the African region is 6.1%. Infection in the neonatal period and in childhood predisposes to chronic infection.1-7 The WHO estimated that chronic hepatitis B resulted in 880 000 deaths from cirrhosis and hepatocellular carcinoma in 2015. Hepatitis B birth dose and universal HBV vaccination, together with third-trimester tenofovir in highly viraemic pregnant women, are highly effective in reducing incident chronic hepatitis B infection in endemic regions. However, the rates of end-stage liver disease in chronically infected persons will remain substantial as a large reservoir of chronic infection remains. This disease burden needs to be progressively eliminated worldwide. The present lack of curative therapy has spurred research into potential targets for novel drug development. Improved understanding of the pathobiology of the virus and the temporal but complex host immunological response is key to understanding the rationale, mechanism of action and appropriate combinations of newer, potentially curative treatments.
Virology
Hepatitis B virus (HBV) is an enveloped 42 nm diameter DNA virus, of the Hepadnaviridae family, containing a partially double-stranded relaxed circular DNA genome of 3 200 base pairs in length. The DNA is replicated via reverse transcription of a RNA pregenome within the capsid during packaging of the RNA and the viral reverse transcriptase. The virion binds to a cellular receptor, sodium taurocholate co-transporting polypeptide (NTCP), and is subsequently internalised into the cell. After entry into the cell, the first event in HBV DNA replication is conversion of the relaxed circular DNA genome into a covalently closed circular DNA (cccDNA) minichromosome in the nucleus of hepatocytes (Fig. 1).
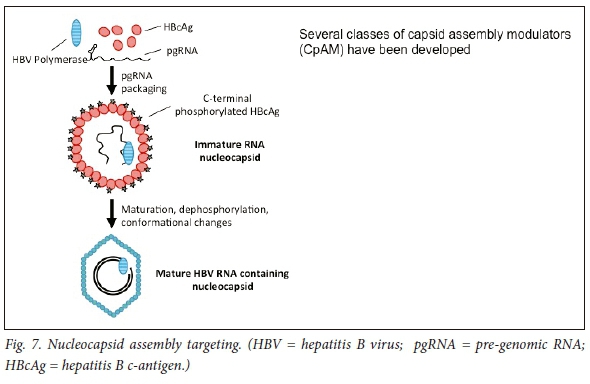
cccDNA is the transcriptional template for transcription of the viral RNAs. cccDNA persists as a minichromosome in the nucleus of the infected cells. Importantly, cccDNA is synthesised from relaxed circular DNA (rcDNA), either from incoming infecting virions or from subsequent intracellular nucleocapsids via an intracellular cccDNA shuttle amplification pathway. The end product of reverse transcription, rcDNA is directed to the host cellular nucleus to form more cccDNA.8,9 Packaging of the viral pre-genomic RNA (pgRNA) occurs as a complex with the HBV reverse transcriptase protein into an icosahedral 30 nm diameter capsid, composed of copies of the viral core. The HBV capsid is assembled into core particles, containing pgRNA and the viral polymerase (reverse transcriptase). Within the nucleocapsid, the pgRNA is reverse transcribed by the viral reverse transcriptase-polymerase to form incomplete rcDNA, and HBV capsids are subsequently coated with hepatitis B surface antigen (HBsAg) to form mature virus particles.10
Complete and incomplete viral particles are thus secreted into the circulation. Empty non-DNA-containing virions are exuded from the cell and can be detected in serum; importantly, recent findings have shown that RNA-containing particles are released. It is noteworthy that inhibition of cccDNA synthesis is not affected by current nucleoside analogue therapy, as only DNA synthesis is targeted. The product of the hepatitis Bx gene (HBx) has been shown to be necessary for the transcription from cccDNA through epigenetic regulation.11,12
HBV DNA fragments are integrated into the genome of hepatocytes but integration is not a requirement for replication of HBV. Hepatitis B e-antigen (HBeAg) is a soluble, dimeric protein that is secreted from hepatocytes. HBeAg is processed from the precore protein. The bulk of amino acids are shared with HBcAg, but HBeAg posseses an N-terminal extension of 10 amino acids and a C-terminal truncation of 34 amino acid residues. Serum HBeAg is associated with high levels of viral infection and can be used to monitor temporal changes and treatment response. HBsAg is also produced following transcription of integrated viral DNA.13 The large envelope glycoprotein on the surface of HBV and hepatitis D virus (HDV) has been demonstrated to play a pivotal role in virus entry. The antigenic loop on the S protein mediates attachment to cell surface heparan sulfate proteoglycans (HSPGs).14,15 There is variation in sequence (up to 12% of nucleotides) between isolates of HBV and up to nine genotypes (A to I) have been described on the basic nucleotide sequence divergence.
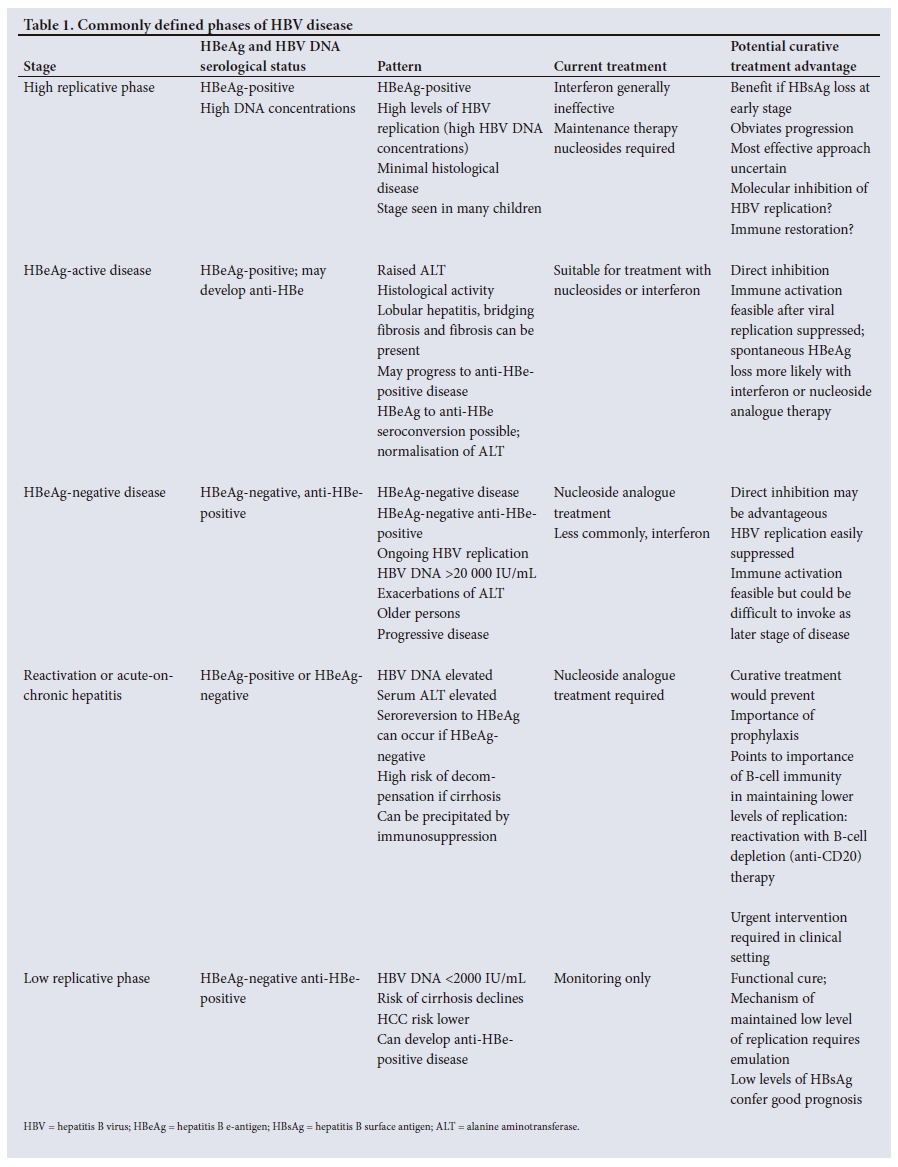
Natural history
The natural history of HBV infection is still being delineated. Most persons infected at birth or childhood develop persistent HBV infection.1-3 Different, age-dependent phases of the disease have long been recognised (Table 1). In the high replicative, low inflammatory phase, HBsAg and HBeAg are detectable in serum, serum HBV DNA concentrations are high, but serum aminotransferases may be only minimally increased or normal. However, recent immunological reports have argued that antigen-specific immune responses exist in a proportion of these so-called immune-tolerant patients and that this phase of the disease is associated with clonal expansion of hepatocytes and viral integration. Adolescents may harbour hepatitis B functionally active specific T cells.16 Serum alanine aminotransferase (ALT) is not a measure of immunological response. However, robust, functional and broadly specific T lymphocyte specificities are lacking, suggesting a degree of functional impairment and clonal tolerance.17 In HBeAg-positive patients, progression to cirrhosis occurs at a rate of 2 - 5.5% per annum with a cumulative 5-year incidence of progression of 8 - 20%. The high replicative phase is followed by active chronic hepatitis with progressive necro-inflammatory disease, or by HBeAg seroconversion and remission in a proportion of patients (the inactive carrier state). Early seroconversion to anti-HBe may lead to remission of the disease and may confer a good prognosis, according to the degree of the liver damage. In HBeAg-positive persons a spontaneous remission is possible in about 10 - 15% of the cases per year. Inactive carriers characteristically manifest persistently normal aminotransferases and HBV DNA concentrations of <2 000 IU/mL. A proportion of HBsAg-positive persons lose HBsAg at a rate of 0. 5 - 2% per year.
HBeAg-negative anti-HBe-positive disease is a common pattern of disease. HBeAg is undetectable in these persons owing to the selection of HBV virions not expressing HBeAg (pre-core mutant HBV). Several mutations in the pre-core region that preclude HBeAg synthesis have been identified in HBeAg-negative carriers, in whom histochemical detection of HBcAg in hepatocytes and histological evidence of active chronic hepatitis is present. Individuals with anti-HBe-positive chronic hepatitis B tend to be older, and have more progressive necro-inflammatory change. HBeAg-negative chronic hepatitis has a variable course, with fluctuating serum aminotransferases and HBV DNA concentrations. Progression to cirrhosis generally occurs more rapidy in anti-HBe-positive disease at a yearly rate of 8 - 20%. Levels of HBsAg and HBV DNA are generally lower than in HBeAg-positive patients.
The reported 5-year cumulative incidence of hepatic decompensation in patients with cirrhosis is 16%. The cumulative probability of survival in 366 cases of HBsAg-positive compensated cirrhosis was 84% and 68% at 5 and 10 years, respectively.18-21 Risk factors associated with a high rate of progression have been identified and include age, male gender, high aminotransferases, high HBV DNA concentrations, high HBsAg levels, genotype C and basal core promoter expression. Higher rates of HCC have been found in individuals with genotypes C and F v. B or D. HCC occurs at a younger age in those with genotypes F, or with subtypes of genotype A found in southern Africa.
HBV and the immune system
The outcome of the HBV infection is determined by the host immune response. Whereas 95% of immunocompetent adults clear the infection, only 5 - 10% of the children resolve the acute infection. Resolution of acute hepatitis B is associated with functionally efficient, broadly specific antiviral T-cell responses, which are preceded by the induction of intracellular innate responses at the early stages of infection.22 Persons resolving HBV infection have vigorous HBV-specific CD4+ and CD8+ T-cell repertoires and function when compared with chronic infection.23,24 Long-lasting protective memory provides persistent control of infection, which is probably sustained by continuous stimulation of the immune system by residual amounts of virus. Chronic virus infection is characterised by a deeply dysfunctional immune response including a lack of protective memory T-cell maturation and an exhausted HBV-specific T-cell response. T-cell exhaustion is thought to be the result of persistent exposure to soluble HBV antigens (HBeAg and HBsAg).25 The liver environment may be tolerogenic. The degree of T-cell impairment is related to the level of HBV replication and antigen load.26,27 Chronic HBV infection is characterised by a low frequency of HBV-specific CD8+ T-cell responses, an impaired production of interleukin (IL-2), an impaired proliferation of CD4+ T cells, an increase in the number of regulating T cells (Treg) in the liver and in the number of IL-10-secreting T cells.28 Although T-cell responses can be detected in young adults, HBV-specific T cells are weak and functionally impaired. The hepatic inflammation is not proportional to the quantity of hepatitis B-specific CD8+ T cells.23,24 Other factors such as chemokines and natural killer cell activation are important. The essential role of B cell immunity in clearance of hepatitis B and reactivation require further study.29 Even at the early phase of HBV DNA integration, clonal expansion hepatocytes indicate that disease is underway.
Current treatment of hepatis B
A large worldwide reservoir of chronic HBV infection remains. The objective of therapy is to prevent progression to cirrhosis, and end-stage liver disease, including HCC. Over the past three decades, conventional interferon alpha pegylated interferon alpha (PEG-IFNcx), nucleotide analogues including lamivudine, adefovir, entecavir, telbivudine, tenofovir and emtricitabine have been shown to delay progression of cirrhosis, reduce but not eliminate the risk of HCC, improve survival, and reduce the need for liver transplantation.30
The REVEAL studies31,32 showed a reduction in HCC risk with lower HBV DNA concentrations. The efficacy of IFNci is restricted. Some pre-treatment and on-treatment factors predictive of response to PEG-IFNc have been identified, including genotype. Successful finite cures of hepatitis B are possible with interferon therapy, but are rare. HBsAg concentrations >20 000 IU/ml at treatment week 12 have a high negative predictive value of unsuccessful treatment (irrespective of HBV genotype) in HBeAg-positive patients.33 The side-effects of interferon treatment mitigated its current use. PEG IFNc added to tenofovir may result in greater suppression of HBsAg than treatment with the nucleoside alone.34 Various combinations of, or sequential therapy with, PEG IFNc and nucleoside analogues reduce HBsAg synthesis, but PEG-IFNc has restricted efficacy (Fig. 2).
HBV DNA replication can be successfully controlled, with nucleos(t)ide analogues that have a high barrier to resistance (entecavir and tenofovir), thus reducing the probability of adverse outcomes, but lifelong therapy is needed for the majority.35 Unfortunately, a large burden of resistant HBV has been engendered in countries where sequential lamivudine and adefovir have been used. The sequential use of entecavir to treat lamivudine resistance increases the risk of entecavir resistance. A switch to tenofovir is effective in patients with confirmed lamivudine, telbivudine, adefovir or entecavir resistance. Nucleos(t)ide analogues with a high barrier to resistance are now recommended as firstline therapy. A reduced incidence of HCC with long-term nucleoside-analogue therapy compared with controls has been reported in cirrhotic patients.36-38
The prospects of a cure of hepatitis B
A functional cure could be characterised as resulting in a sustained loss of HBsAg, which is associated with improved clinical outcomes. A complete sterilising cure with undetectable HBsAg in serum and eradication of intrahepatic cccDNA, as well as integrated cccDNA, is not presently feasible. Also, loss of HBsAg later in life after the development of cirrhosis will not eliminate the risk of HCC. New molecular therapeutics and immunological strategies are emerging, but the stable and resistant reservoir of episomal cccDNA and a dysfunctional or ineffective immune response impedes cure at present. Combinations of, or sequential therapy with, PEG IFNc and nucleoside analogues reduce HBsAg synthesis, but the effects on cure are limited.39,40
Nucleoside analogues compete at the HBV polymerase catalytic site during synthesis of nascent HBV DNA, preventing the formation of a covalent bond with the adjoining nucleotide, and thus causing chain termination of the elongating DNA.
Nucleosides suppress cccDNA amplification but do not act on preformed cccDNA.41 The stable maintenance and function of cccDNA pools in the nuclei of infected cells provides a continued source of viral RNA transcripts and generally precludes curing with nucleoside analogue therapy and nessitates lifelong therapy.
Finite therapywith nucleosides or interferon can result in HBsAg loss in some patients42,43 and may be predicted by HBsAg levels in HBeAg negative patients.44 Consolidation therapy after HBsAg loss is advisable.45 Long-term (more than 4-year) entecavir or tenofovir therapy can be discontinued for non-cirrhotic HBeAg-negative patients if close follow-up is possible, as the cumulative rates of virological relapse are high at 67% and 31%, respectively, with serum HBV DNA cutoff values of >20 000 IU/mL and >2 000 IU/L, respectively. The probability of relapse seems to diminish 6 months after cessation of treatment.46,47 There would be several potential benefits of finite therapy with nucleos(t)ide analogues, including improved adherence and cost savings for healthcare systems.
Potential new treatments
Several recent reviews have summarised prospective treatments.48-50 A full review of potential curative strategies is beyond the scope of this article, but prospective new compounds are reviewed in two recent publications.51,52 However, cure of hepatitis B is the next goal of therapy of hepatitis B (Fig. 3).
Tenofovir alafenamide fumarate (TAF) is an oral phosphonoamidate prodrug of tenofovir. Uptake of the parent nucleotide and its active diphosphate metabolite into lymphoid cells and hepatocytes is enhanced due to improved plasma stability and a different intracellular activation mechanism for TAF.
Similar rates of viral suppression but higher rates of ALT normalisation with TAF have been reported. Switching to TAF results in improvements in bone density and renal function.53,54 Similarly, TAF has a high barrier to resistance but is not likely to improve rates of cure.55-57 Other nucleosides in the pipeline include besifovir.58
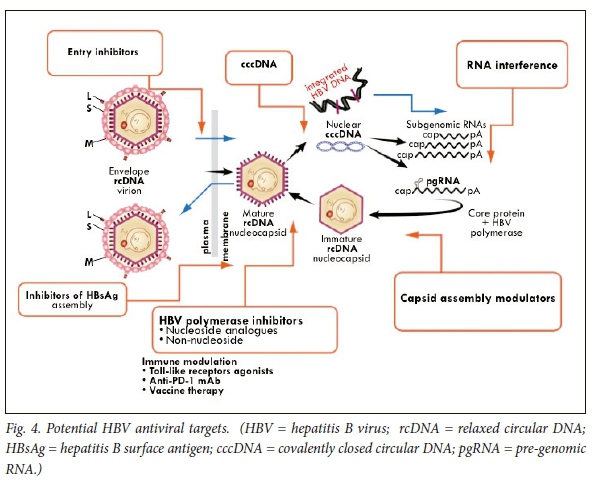
Several new inhibitory molecules are being investigated. These include entry inhibitors, short interfering RNA (siRNAs), or capsid inhibitors (Fig. 4).49
Entry inhibitors
The sodium taurocholate co-transporting polypeptide has recently been recognised as the HBV (and HDV) receptor.59 A synthesised acylated pre-S peptide derivative of the large HBsAg protein blocks entry of HBV in experimental cells (Myrcludex B; MYR GmbH, Burgwedel) and is currently being studied in chronic HBV and HDV infection.60 Encouraging phase 1 and 2 studies with nucleosides and interferon are in progress. Parenterally administered neutralising antibodies are a potential alternative approach.
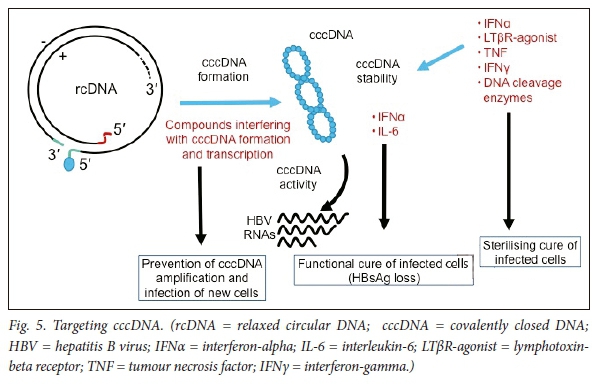
Targeting cccDNA
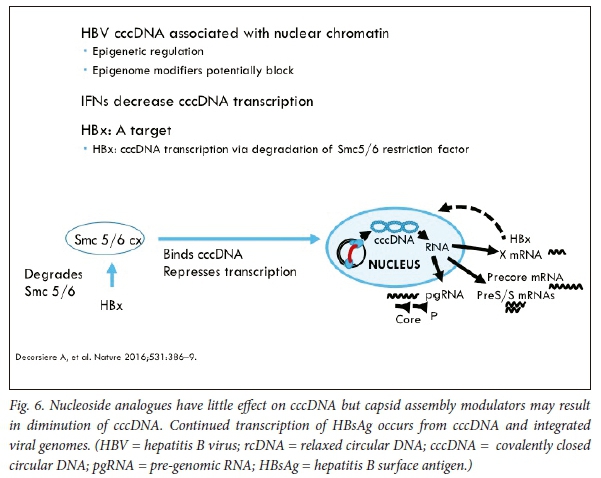
Relaxed, unwound chromatin is associated with the activation of gene expression and, in contrast, compacted chromatin is associated with repression of gene expression. Epigenetic regulation of transcription could potentially disrupt transcription from cccDNA. cccDNA contains methylation-prone CpG islands61 and thus epigenetic silencing of cccDNA transcription may be possible.62-65 Two classes of enzymes, histone deacetylases (HDACs) and histone acetyltransferases (HATs), regulate the acetylation status of histones; post-translational modification of histones by acetylation, phosphorylation, methylation and ubiquitylation reactions could potentially be targeted (Fig. 5).
cccDNA within infected hepatocytes could be degraded. HBx is required for cccDNA transcription and viral replication (via degradation of the Smc5/6 restriction factor) and is an attractive target (Fig. 6).66 Assays to detect cccDNA have, unfortunately, not been standardised. Lymphotoxin-β receptor signalling has been shown to activate upregulation of cytidine deaminases.67 Lucifora et al.67 have shown that IFNcx induces specific degradation of the nuclear viral DNA; their experimental data suggest that IFNcx activation and activation of the lymphotoxin-β receptor upregulates APOBEC3A and APOBEC3B, two cytidine deaminases, in HBV-infected cells, primary hepatocytes and human liver tissue specimens, to degrade cccDNA.67 DNA cleavage enzymes, including CRISPR-associated nucleases, are being tested in experimental models. Mutational inactivation of HBV DNA by clustered, short palindromic repeats (CRISPRs) and CRISPR-associated (Cas) proteins is a powerful tool to potentially induce cleavage of specific HBV DNA targets, after direction by a synthetic guide RNA base-paired to the target HBV DNA sequence.68,69 The CRISPR/Cas9 system has been shown to diminish production of HBV capsid and surface proteins in Huh-7 transfected cells. Recently Seeger et al.70 established HepG2 cells expressing the NTCP receptor that are susceptible to HBV infection. Guide RNAs inhibited HBcAg expression in HepG2 cells, which showed that CRISPR/Cas9 mechanisms for efficient inactivation of viral genes targeting cccDNA may be possible. Potential off-target effects will need to be addressed before they can be tested in clinical trials.
Targeting viral transcripts
Antisense, siRNA targeting is being tested. siRNA can be engineered to target HBsAg transcripts and subsequent degradation of the mRNA. Several antisense siRNA formulations are under clinical or preclinical evaluation. ARC 520 is a RNA interference (RNAi)-based, liver-targeted antiviral RNA comprising a combination of two cholesterol-conjugated siRNA molecules that directly target HBV RNA transcripts to trigger sequence-specific knock-down modulation of gene expression, and thus reduce HBV DNA and viral proteins.71-76
Prolonged RNAi therapy with ARC-520 in treatment-naive, HBeAg-positive and HBeAg-negative patients with chronic HBV results in significant reductions in HBsAg.75 The reduction in HBeAg-positive patients is greater than in HBeAg-negative patients, which probably reflects reductions in HBsAg from cccDNA in the former v. integrated DNA in HBeAg negative patients. A phase 2a study evaluating the multi-dose activity of ARB-1467 in HBeAg-positive and HBeAg-negative virally suppressed patients with HBV has been reported. ARB-1467 is given by injection (2 mg/mL) IV over 2 hours for 3 months; pre-medications (including steroids) were given to mitigate infusion-related adverse events. Significant reductions in HBsAg with single doses of ARB-1467 0.2 mg/kg and 0.4 mg/kg were noted with stepwise, additive reductions with multiple doses. There were no significant differences in serum HBsAg between HBeAg-negative and HBeAg-positive patients.77 Currently a potential limitation is the need for intravenous administration, the possibility of off-target binding and the potential toxicity of the excipient. Studies are required to determine whether knockdown of HBsAg will restore antigen-specific immune responsiveness.78
Nucleic acid polymers are oligonucleo-tides with antiviral activity but which are not sequence dependent. Several phase II clinical trials, including in combination with interferon, are in progress and have demonstrated a profound decline in serum HBsAg concentrations. The compounds under investigation, REP 2139-Mg or REP 2165-Mg used in combination with tenofovir and PEG-IFNa and given by intravenous infusion, block the entry and formation of subviral particles by interfering with apolipoprotein-HBsAg interactions.52,79,80
The selective effect of REP 2139-Mg on serum HBsAg is consistent with the targeting of subviral particle release in cells.
Capsid inhibitors
Because of the pivotal interaction between the HBV nucleocapsid and cccDNA, the HBV capsid is a crucial potential target. HBV replication is in part regulated by the kinetics of assembly of the nucleocapsid particle, which comprises the viral capsid proteins, HBV polymerase and pre-genomic RNA (reviewed by Berke et al.).'811Thus inhibitors of either encapsidation, or compounds that result in capsid dissembly, impede the entry of rcDNA into the nucleus and could thus inhibit transformation of rcDNA to cccDNA. A number of compounds active against the HBV core are in development. Capsid assembly modulators (CAMs) hasten the kinetics of the HBV nucleocapsid complex assembly and thereby prevent the encapsidation of the polymerase-pgRNA composite and block the reverse transcription of the pgRNA. Moreover, HBV core protein regulates cccDNA transcription and replication steps, perhaps via reducing acetylation of cccDNA-bound histones (Fig. 7). Thus, HBsAg levels may be reduced (Fig. 2).
Several compound classes have been reported to disrupt polymerase-pgRNA encapsidation at the nucleation step, resulting in either empty or intact capsids (a class I mechanism of action) or alternatively affect the formation of pleiomorphic noncapsid structures referred to as class II compounds.
Recently, some capsid inhibitors have also been found to reduce intrahepatic cccDNA concentrations.82 AB-423 (a capsid inhibitor) misdirects capsid assembly and thus inhibits pgRNA encapsidation. AB-423 exhibits a dual mode of action in C3AhNTCP cells, namely inhibition of encapsidation of pgRNA during ongoing infection in addition to inhibition of rcDNA-cccDNA synthesis, presumably via inhibition of the capsid uncoating step. Second-generation RNA interference agents that primarily target HBsAg production are in development. In a HBV-infected chimeric mouse model an additive benefit of combination therapy of ARB-423 and ARB-1740, and further benefits with PEG-IFNcx and entecavir have been demonstrated. Reduced cccDNA transcriptional activity was demonstrable.83
The capsid assembly modulator JNJ-56136379 prevents de novo infection of primary human hepatocytes (PHH) with hepatitis B virus. The compound induces the formation of empty HBV capsids with normal geometry and size. The compound is active across a broad panel of clinical isolates and is potentially additive to synergistic with nucleos(t)ide analogues. The compound may also block cccDNA formation in PHHs, resulting in the reduction of HBV RNA and antigens.84,85
The combination of GLS4JHS and ritonavir was well tolerated and produced a rapid and substantial decrease in HBV-DNA levels in patients chronically infected with HBV. 86
Regulation of immunity
Restoration of adaptive immunity remains a challenge. Chronic hepatitis B is associated with several identifiable levels of immune dysfunction, including functional exhaustion of HBV-specific CD8+ T cells, as a consequence of exposure to abundant quantities of HBsAg and HBeAg, expression of inhibitory molecules such as programmed cell death protein 1(PD-1), and consequently the loss of antigen-specific T cell effector function. Blocking expression of inhibitory regulatory receptors (checkpoint inhibition) could possibly restore functional T-cell activity to exhausted lymphoid cells. Blockade of the PD-1 pathway with woodchuck anti-programmed death ligand 1 (PD-L1 antibody), therapeutic DNA vaccination, and treatment with entecavir, as well as enhanced virus-specific T cell immunity, has led to resolution of chronic infection in woodchucks.87
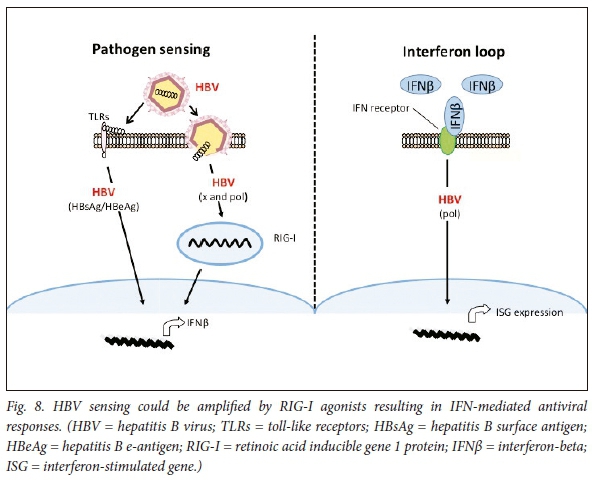
Experimental toll-like receptor (TLR) agonists suggest that HBV replication could be inhibited by the activation of innate immune responses in the liver. GS-9620, is an oral agonist of TLR-7. The immunological and pharmacodynamic effects are being studied in humans.88 A dose-dependent induction of peripheral interferon-stimulated gene 15 (ISG15) gene expression has been observed, but without a clinically significant change in HBsAg concentrations.89
Recently Pallett et al.90 demonstrated that myeloid-derived suppressor cells suppress T cell-mediated immunopathology in chronic hepatitis B: these cells expanded transiently in acute resolving hepatitis B infection but decrease in frequency prior to peak hepatic injury. During chronic infection, arginase-expressing myeloid-derived suppressor cells and circulating arginase increased in phase with HBV replication without immunopathology, and thus L-arginine decreased. The myeloid-derived suppressor cells potently inhibited T cells in a partially arginase-dependent manner, demonstrating the possible potential of the arginase-expressing suppressor cells to regulate liver immunopathology during hepatitis B.
Other novel molecular therapeutic strategies and immunological treatments include the possible application of therapeutic vaccines to boost HBV-specific T-cell responses or alter the dysfunctional immune response. Tarmogens, i.e. yeast-based immunotherapy platforms, have been used to make vaccine candidates expressing HBV X, S, and core antigens.91 GS4774 is a genetically modified yeast organism engineered to express HBV antigens to activate T cells. Other possible therapeutic vaccines, including adenovirus fusion proteins, are currently being evaluated in clinical trials.
SB 9200 is a unique oral prodrug of the dinucleotide SB9000, and acts as a retinoic acid-inducible gene 1 (RIG-I) activator. The drug is thought to bind and activate RIG-I, thus increasing sensitivity to viral proteins, resulting in IFN-mediated antiviral immune responses in virus-infected cells, as well as to inhibit the packing reaction during HBV encapsidation (Fig. 8).
New markers of HBV infection
It is difficult to measure or monitor intrahepatic cccDNA levels or its transcriptional activity; testing requires the use of liver tissues, or surrogate markers in serum that accurately reflect infection through different phases of the disease. Various HBV particles released into peripheral blood are of increasing interest as surrogates to monitor cccDNA or transcriptional activity.
HBV RNA
It has become apparent that HBV may secrete, at low levels, RNA-containing particles. Numerous recent reports have demonstrated the detection of HBV RNA in serum samples of HBV-infected patients at0.1 - 1% of HBV DNA levels, in the absence of antiviral treatment. The nature of the HBV RNA in the serum remains to be clarified. Some reports suggest that the RNA could be original pgRNA but it is not known whether these HBV RNA virions are infectious. There are technical challenges in discriminating HBV RNA with a large excess of rcDNA that is structurally related to CCC.92
Since the DNA-containing virions are present at high concentrations, routine DNase digestion, to remove the viral DNA before RNA detection is required to completely remove all viral DNA. Serum HBV RNA is being assessed as a surrogate to monitor CCC DNA. When viral DNA synthesis is effectively suppressed with antiviral therapy, a rapid RNA decrease early during therapy with chain terminators may predict viral clearance or, equally, the persistence of serum HBV RNA might predict viral rebound after stopping treatment.
Quantitative measurements of serum HBsAg may have prognostic significance. However, expression of HBsAg may be the predominant source of serum HBsAg, which may explain the loss of correlation between serum HBsAg levels and hepatic cccDNA, in later stages of HBV infection.
Hepatitis B core-related protein
Hepatitis B core-related protein (HBcrAg) has been identified as an aberrantly processed pre-core protein comprising the entire pre-core region including the uncleaved signal peptide (a 29-residue N-terminal extension relative to HBc but similarly to HBeAg, devoid of the C-terminal arginine-rich domain which mediates HBV RNA and DNA binding by HBcAg (reviewed by Hu and Hu).92
HBcrAg in serum samples has been tested in a number of clinical studies as a potential surrogate for monitoring cccDNA. Generally 'HBcrAg' has been used to designate a combination of HBc and HBe and p22cr. HBcrAg concentrations correlate well with serum HBV DNA and HBsAg levels, and with HBcAg-positive hepatocytes.
Conclusion
Current therapies with nucleoside analogues achieve remission in most patients. Nucleoside analogues can be stopped in a proportion of patients. The majority of patients treated with nucleoside analogues require maintenance therapy and although these therapies have reduced morbidity and mortality from the disease, the majority are not cured.
It is unclear whether all cccDNA chromatin would need to be eradicated for a cure of hepatitis B, or whether lowering levels to a lower threshold could reduce HBV replication and hence disease progression (as may be the case in inactive carriers). Unfortunately, there are no definitively proven serological surrogates for cccDNA for study in pilot phase experiments - these trials in patients may require liver biopsy to quantify tissue cccDNA. HBsAg quantitation may serve this purpose but phase 1 dose-finding studies may also require precise quantitation and characterisation of cccDNA in liver tissue.93,94
Cures for hepatitis B will no doubt require multiple synergistic therapies to achieve either eradication from the host, or alternatively a functional cure. The complex dysfunctional immune response will require further understanding to remedy. Curative regimens will possibly demand a complex combination or sequence of agents, to result in viral suppression via nucleoside analogue treatment, selective cccDNA inhibitors or modifiers to deplete silence or degrade cccDNA, and restoration of the exhausted T-cell repertoire, as well as perhaps, agents to block the entry of HBV into the hepatocyte. Nonetheless a number of promising lines of treatment are being examined and regulatory pathways are being defined.95
It is not yet clear at what stages of the disease and to what extent the dysfunctional adaptive immune response apparent in hepatitis B infection can be restored, and to what extent exhaustion of cytolytic T cells induced by high levels of exposure to hepatitis B antigens can be reduced by a combination of suppression of viral replication as well as blockade of inhibitory antibody. Neither are the risks of severe uncontrolled T-cell cytolytic responses fully ascertained. Painstaking empirical data are required. The correct sequencing and combinations of treatment with either host or viral targeting agents have yet to be established.
Many safety considerations exist. Nucleic acid-based gene knockdown and silencing molecules will need to achieve precise sequence specificity and be devoid of off-target biological interactions with proteins other than the targeted gene transcripts.
The management of chronic hepatitis B has advanced significantly, and treatments that effectively suppress the virus with no risk of resistance need to be widely applied. New agents to eliminate active replicative HBV infection or achieve a functional cure are being avidly sought. Their incorporation into strategies to optimise combination therapies will be tested over the next decade.
Acknowledgements. None.
Author contributions. Sole author.
Funding. GD has received grant support and advisory board fees from: AbbVie, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Merck Sharp Dohme and Roche.
Conflicts of interest. None.
References
1. Beasley RP, Hwang LY, Lee GC, et al. Prevention of perinatally transmitted hepatitis B virus infections with hepatitis B immune globulin and hepatitis B vaccine. Lancet 1983;2(8359):1099-1102. [ Links ]
2. Beasley RP, Hwang LY, Lin CC, et al. Incidence of hepatitis B virus infections in preschool children in Taiwan. J Infect Dis 1982;146(2):198-204. [ Links ]
3. McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985;151(4):599-603. [ Links ]
4. Gish RG, Cohen CA, Block JM, et al. Data supporting updating estimates of the prevalence of chronic hepatitis B and C in the United States. Hepatology 2015;62(5):1339-1341. https://doi.org/10.1002/hep.28026 [ Links ]
5. Zhang W, Ji Z, Wang L, Xiao D, Yan Y. A meta-analysis of HBsAg-positive rate among general Chinese populations aged 1 - 59 years. Infect Dis 2015;47(12):878-888. https://doi.org/10.3109/23744235.2015.1064541 [ Links ]
6. Lok AS. Hepatitis B: 50 years after the discovery of Australia antigen. J Viral Hepat 2016;23(1):5-14. https://doi.org/10.1111/jvh.12444 [ Links ]
7. Liu J, Zhang S, Wang Q, et al. Seroepidemiology of hepatitis B virus infection in 2 million men aged 2149 years in rural China: A population-based, cross-sectional study. Lancet Infect Dis 2016;16(1):80-86. https://doi.org/10.1016/S1473-3099(15)00218-2 [ Links ]
8. Bock CT, Schranz P, Schroder CH, Zentgraf H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994;8(3):215-229. [ Links ]
9. Bock CT, Schwinn S, Locarnini S, et al. Structural organization of the hepatitis B virus minichromosome. J Mol Biol 2001;307(1):183-196. https://doi.org/10.1006/jmbi.2000.4481 [ Links ]
10. Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res 2015;121:82-93. https://doi.org/10.1016/j.antiviral.2015.06.020 [ Links ]
11. Belloni L, Pollicino T, De Nicola F, et al. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci USA 2009;106(47):19975-19979. https://doi.org/10.1073/pnas.0908365106 [ Links ]
12. Lucifora J, Arzberger S, Durantel D, et al. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol 2011;55(5):996-1003. https://doi.org/10.1016/j.jhep.2011.02.015 [ Links ]
13. Marion PL, Salazar FH, Alexander JJ, Robinson WS. State of hepatitis B viral DNA in a human hepatoma cell line. J Virol 1980;33(2):795-806. [ Links ]
14. Le Duff Y, Blanchet M, Sureau C. The pre-S1 and antigenic loop infectivity determinants of the hepatitis B virus envelope proteins are functionally independent. J Virol 2009;83(23):12443-12451. https://doi.org/10.1128/JVI.01594-09 [ Links ]
15. Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007;46(6):1759-1768. https://doi.org/10.1002/hep.21896 [ Links ]
16. Mason WS, Gill US, Litwin S, et al. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis b patients considered immune tolerant. Gastroenterology 2016;151(5):986-998. https://doi.org/10.1053/j.gastro.2016.07.012 [ Links ]
17. Bertoletti A, Kennedy PT. The immune tolerant phase of chronic HBV infection: New perspectives on an old concept. Cell 2014;12(3):258-263. https://doi.org/10.1038/cmi.2014.79 [ Links ]
18. Fattovich G. Natural history and prognosis of hepatitis B. Semin Liver Dis 2003;23(1):47-58. https://doi.org/10.1055/s-2003-37590 [ Links ]
19. Fattovich G, Giustina G, Realdi G, Corrocher R, Schalm SW. Long-term outcome of hepatitis B e antigen-positive patients with compensated cirrhosis treated with interferon alfa. European Concerted Action on Viral Hepatitis (EUROHEP). Hepatology 1997;26(5):1338-1342. https://doi.org/10.1002/hep.510260536 [ Links ]
20. Fattovich G, Giustina G, Schalm SW, et al. Occurrence of hepatocellular carcinoma and decompensation in western European patients with cirrhosis type B. Hepatology 1995;21(1):77-82. [ Links ]
21. Fattovich G, Olivari N, Pasino M, D'Onofrio M, Martone E, Donato F. Long-term outcome of chronic hepatitis B in Caucasian patients: Mortality after 25 years. Gut 2008;57(1):84-90. https://doi.org/10.1136/gut.2007.128496 [ Links ]
22. Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science 1999;284(5415):825-829. [ Links ]
23. Maini MK, Boni C, Lee CK, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med 2000;191(8):1269-1280. [ Links ]
24. Webster GJ, Reignat S, Brown D, et al. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: Implications for immunotherapy. J Virol 2004;78(11):5707-5719. https://doi.org/10.1128/JVI.78.11.5707-5719.2004 [ Links ]
25. Kondo Y, Ninomiya M, Kakazu E, Kimura O, Shimosegawa T. Hepatitis B surface antigen could contribute to the immunopathogenesis of hepatitis B virus infection. ISRN Gastroenterol 2013;2013:935295. https://doi.org/10.1155/2013/935295 [ Links ]
26. Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol 2005;5(3):215-229. https://doi.org/10.1038/nri1573 [ Links ]
27. Protzer U, Maini MK, Knolle PA. Living in the liver: Hepatic infections. Nat Rev Immunol 2012;12(3):201-213. https://doi.org/10.1038/nri3169 [ Links ]
28. Bertoletti A, Maini MK, Ferrari C. The host-pathogen interaction during HBV infection: immunological controversies. Antivir Ther 2010;15(Suppl 3):15-24. https://doi.org/10.3851/IMP1620 [ Links ]
29. Thursz M. Basis of HBV persistence and new treatment options. Hepatology Int 2014;8(Suppl 2):486-491. https://doi.org/10.1007/s12072-013-9504-6 [ Links ]
30. Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Eng J Med 2004;351(15):1521-1531. https://doi.org/10.1056/NEJMoa033364 [ Links ]
31. Lee MH, Yang HI, Liu J, et al. Prediction models of long-term cirrhosis and hepatocellular carcinoma risk in chronic hepatitis B patients: Risk scores integrating host and virus profiles. Hepatology 2013;58(2):546-554. https://doi.org/10.1002/hep.26385 [ Links ]
32. Chen CF, Lee WC, Yang HI, et al. Changes in serum levels of HBV DNA and alanine aminotransferase determine risk for hepatocellular carcinoma. Gastroenterology 2011;141(4):1240-1248. https://doi.org/10.1053/j.gastro.2011.06.036 [ Links ]
33. Sonneveld MJ, Rijckborst V, Boucher CA, Hansen BE, Janssen HL. Prediction of sustained response to peginterferon alfa-2b for hepatitis B e antigen-positive chronic hepatitis B using on-treatment hepatitis B surface antigen decline. Hepatology 2010;52(4):1251-1257. https://doi.org/10.1002/hep.23844 [ Links ]
34. Brouwer WP, Xie Q, Sonneveld MJ, et al. Adding pegylated interferon to entecavir for hepatitis B e antigen-positive chronic hepatitis B: A multicenter randomized trial (ARES study). Hepatology 2015;61(5):1512-1522. https://doi.org/10.1002/hep.27586 [ Links ]
35. Seto WK, Lau EH, Wu JT, et al. Effects of nucleoside analogue prescription for hepatitis B on the incidence of liver cancer in Hong Kong: a territory-wide ecological study. Alimentary Pharmacol Ther 2017;45(4):501-509. https://doi.org/10.1111/apt.13895 [ Links ]
36. Hosaka T, Suzuki F, Kobayashi M, et al. Long-term entecavir treatment reduces hepatocellular carcinoma incidence in patients with hepatitis B virus infection. Hepatology 2013;58(1):98-107. [ Links ]
37. Singal AK, Salameh H, Kuo YF, Fontana RJ. Meta-analysis: the impact of oral anti-viral agents on the incidence of hepatocellular carcinoma in chronic hepatitis B. Alimentary Pharmacol Ther 2013;38(2):98-106. https://doi.org/10.1111/apt.12344 [ Links ]
38. Coffin CS, Rezaeeaval M, Pang JX, et al. The incidence of hepatocellular carcinoma is reduced in patients with chronic hepatitis B on long-term nucleos(t)ide analogue therapy. Alimentary Pharmacol Ther 2014;40(11-12):1262-1269. https://doi.org/10.1111/apt.12990 [ Links ]
39. Wong GL, Wong VW, Chan HL. Combination therapy of interferon and nucleotide/nucleoside analogues for chronic hepatitis B. J Viral Hepat 2014;21(12):825-834. https://doi.org/10.1111/jvh.12341 [ Links ]
40. Chan H, Ahn WL, Chuang AJ, Hui F, Tabak R, Mehta G. Predictors of clinical response: results from a a large randomized controlled study with tenofovir disoproxil fumarate plus interferon alpha 2a (PEG) combination for chronic hepatitis B. J Hepatol 2015;62:S235-S262. [ Links ]
41. Dusheiko G. Treatment of HBeAg positive chronic hepatitis B: interferon or nucleoside analogues. Liver Int 2013;33(Suppl 1):137-150. https://doi.org/10.1111/liv.12078 [ Links ]
42. Chen CH, Chiu YC, Lu SN, et al. Serum hepatitis B surface antigen levels predict treatment response to nucleos(t)ide analogues. World J Gastroenterol 2014;20(24):7686-7695. https://doi.org/10.3748/wjg.v20.i24.7686 [ Links ]
43. Zhang Y, Hu XY, Zhong S, et al. Entecavir vs lamivudine therapy for naive patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. World J Gastroenterol 2014;20(16):4745-4752. https://doi.org/10.3748/wjg.v20.i16.4745 [ Links ]
44. Hadziyannis SJ, Sevastianos V, Rapti I, Vassilopoulos D, Hadziyannis E. Sustained responses and loss of HBsAg in HBeAg-negative patients with chronic hepatitis B who stop long-term treatment with adefovir. Gastroenterology 2012;143(3):629-636. https://doi.org/10.1053/j.gastro.2012.05.039 [ Links ]
45. Chi H, Hansen BE, Yim C, et al. Reduced risk of relapse after long-term nucleos(t)ide analogue consolidation therapy for chronic hepatitis B. Aliment Pharmacol Ther 2015;41(9):867-876. https://doi.org/10.1111/apt.13150 [ Links ]
46. Papatheodoridis GV, Idilman R, Dalekos GN, et al The risk of hepatocellular carcinoma is decreasing after the first 5 years of entecavir or tenofovir in Caucasians with chronic hepatitis B. Hepatology 2017;66(5):1444-1453. https://doi.org/10.1002/hep.29320 [ Links ]
47. Papatheodoridis G, Vlachogiannakos I, Cholongitas E, et al. Discontinuation of oral antivirals in chronic hepatitis B: A systematic review. Hepatology 2016;63(5):1481-1492. https://doi.org/10.1002/hep.28438 [ Links ]
48. Zoulim F, Durantel D. Antiviral therapies and prospects for a cure of chronic hepatitis B. Cold Spring Harb Perspect Med 2015;5(4):a021501-a021501. https://doi.org/10.1101/cshperspect.a021501 [ Links ]
49. Kapoor R, Kottilil S. Strategies to eliminate HBV infection. Future Virol 2014;9(6):565-585. https://doi.org/10.2217/fvl.14.36 [ Links ]
50. Gish RG, Given BD, Lai CL, et al. Chronic hepatitis B: Virology, natural history, current management and a glimpse at future opportunities. Antiviral Res 2015;121:47-58. https://doi.org/10.1016/j.antiviral.2015.06.008 [ Links ]
51. Liang TJ, Block TM, McMahon BJ, et al. Present and future therapies of hepatitis B: From discovery to cure. Hepatology 2015;62(6):1893-1908. https://doi.org/10.1002/hep.28025 [ Links ]
52. Block TM, Rawat S, Brosgart CL. Chronic hepatitis B: A wave of new therapies on the horizon. Antiviral Res 2015;121:69-81. https://doi.org/10.1016/j.antiviral.2015.06.014 [ Links ]
53. Buti M, Gane E, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of patients with HBeAg-negative chronic hepatitis B virus infection: A randomised, doubleblind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol 2016;1(3):196-206. https://doi.org/10.1016/S2468-1253(16)30107-8 [ Links ]
54. Chan HL, Fung S, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg-positive chronic hepatitis B virus infection: A randomised, double-blind, phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol 2016;1(3):185-195. https://doi.org/10.1016/S2468-1253(16)30024-3 [ Links ]
55. Bam RA, Birkus G, Babusis D, Cihlar T, Yant SR. Metabolism and antiretroviral activity of tenofovir alafenamide in CD4 T-cells and macrophages from demographically diverse donors. Antivir Ther 2014;19(7):669-677. https://doi.org/10.3851/IMP2767 [ Links ]
56. Bam RA, Yant SR, Cihlar T. Tenofovir alafenamide is not a substrate for renal organic anion transporters (OATs) and does not exhibit OAT-dependent cytotoxicity. Antivir Ther 2014;19(7):687-692. https://doi.org/10.3851/IMP2770 [ Links ]
57. Sax PE, Zolopa A, Brar I, et al. Tenofovir alafenamide vs. tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: A randomized phase 2 study. J Acquir Immune Defic Syndr 2014;67(1):52-58. https://doi.org/10.1097/QAI.0000000000000225 [ Links ]
58. Lai CL, Ahn SH, Lee KS, et al. Phase IIb multicentred randomised trial of besifovir (LB80380) versus entecavir in Asian patients with chronic hepatitis B. Gut 2014;63(6):996-1004. https://doi.org/10.1136/gutjnl-2013-305138 [ Links ]
59. Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012;1:e00049. https://doi.org/10.7554/eLife.00049 [ Links ]
60. Yan H, Liu Y, Sui J, Li W. NTCP opens the door for hepatitis B virus infection. Antiviral Res 2015;121:24-30. https://doi.org/10.1016/j.antiviral.2015.06.002 [ Links ]
61. Zhang Y, Mao R, Yan R, et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by cpg methylation during chronic infection. PLoS one 2014;9(10):e110442. https://doi.org/10.1371/journal.pone.0110442 [ Links ]
62. Vivekanandan P, Thomas D, Torbenson M. Methylation regulates hepatitis B viral protein expression. J Infect Dis 2009;199(9):1286-1291. https://doi.org/10.1086/597614 [ Links ]
63. Guo Y, Li Y, Mu S, Zhang J, Yan Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J Med Virol 2009;81(7):1177-1183. https://doi.org/10.1002/jmv.21525 [ Links ]
64. Kim JW, Lee SH, Park YS, et al. Replicative activity of hepatitis B virus is negatively associated with methylation of covalently closed circular DNA in advanced hepatitis B virus infection. Intervirology 2011;54(6):316-325. https://doi.org/10.1159/000321450 [ Links ]
65. Pollicino T, Belloni L, Raffa G, et al. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006;130(3):823-837. https://doi.org/10.1053/j.gastro.2006.01.001 [ Links ]
66. Decorsiere A, Mueller H, van Breugel PC, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016;531(7594):386-389. https://doi.org/10.1038/nature17170 [ Links ]
67. Lucifora J, Xia Y, Reisinger F, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014;343(6176):1221-1228. https://doi.org/10.1126/science.1243462 [ Links ]
68. Lin SR, Yang HC, Kuo YT, et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Molecular Ther Nucleic Acids 2014;3:e186. https://doi.org/10.1038/mtna.2014.38 [ Links ]
69. Schiffer JT, Swan DA, Stone D, Jerome KR. Predictors of hepatitis B cure using gene therapy to deliver DNA cleavage enzymes: A mathematical modeling approach. PLoS Comp Bio 2013;9(7):e1003131. https://doi.org/10.1371/journal.pcbi.1003131 [ Links ]
70. Seeger C, Sohn JA. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic Acids 2014;3:e216. https://doi.org/10.1038/mtna.2014.68 [ Links ]
71. Wooddell CI, Rozema DB, Hossbach M, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther 2013;21(5):973-895. https://doi.org/10.1038/mt.2013.31 [ Links ]
72. Ivacik D, Ely A, Ferry N, Arbuthnot P. Sustained inhibition of hepatitis B virus replication in vivo using RNAi-activating lentiviruses. Gene Ther 2014;22(2):163-171. https://doi.org/10.1038/gt.2014.94 [ Links ]
73. Mowa MB, Crowther C, Ely A, Arbuthnot P. Inhibition of hepatitis B virus replication by helper dependent adenoviral vectors expressing artificial anti-HBV pri-miRs from a liver-specific promoter. Biomed Res 2014;2014:718743. https://doi.org/10.1155/2014/718743 [ Links ]
74. Watanabe T, Hatakeyama H, Matsuda-Yasui C, et al. In vivo therapeutic potential of Dicer-hunting siRNAs targeting infectious hepatitis C virus. Sci Rep 2014;4:4750. https://doi.org/10.1038/srep04750 [ Links ]
75. Gish RG, Yuen MF, Chan HL, et al. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antiviral Res 2015;121:97-108. https://doi.org/10.1016/j.antiviral.2015.06.019 [ Links ]
76. Sebestyen MG, Wong SC, Trubetskoy V, Lewis DL, Wooddell CI. Targeted in vivo delivery of siRNA and an endosome-releasing agent to hepatocytes. Methods Mol Bio 2015;1218:163-186. https://doi.org/10.1007/978-1-4939-1538-5_10 [ Links ]
77. Seto WK, Yuen MF. New pharmacological approaches to a functional cure of hepatitis B. Clinic Liver Dis 2016;8(4):83-88. https://doi.org/10.1002/cld.577 [ Links ]
78. Durantel D. New treatments to reach functional cure: Virological approaches. Best Pract Res Clin Gastroenterol 2017;31(3):329-336. https://doi.org/10.1016/j.bpg.2017.05.002 [ Links ]
79. Noordeen F, Vaillant A, Jilbert AR. Nucleic acid polymers prevent the establishment of duck hepatitis B virus infection in vivo. Antimicrob Agents Chemother 2013;57(11):5299-5306. https://doi.org/10.1128/AAC.01005-13 [ Links ]
80. Al-Mahtab M, Bazinet M, Vaillant A. Safety and efficacy of nucleic acid polymers in monotherapy and combined with immunotherapy in treatment-naive Bangladeshi patients with HBeAg+ chronic hepatitis B infection. PLoS ONE 2016;11(6):e0156667. https://doi.org/10.1371/journal.pone.0156667 [ Links ]
81. Berke JM, Dehertogh P, Vergauwen K, et al. Capsid assembly modulators have a dual mechanism of action in primary human hepatocytes infected with hepatitis B virus. Antimicrob Agents Chemother 2017;61(8):e00560. https://doi.org/10.1128/AAC.00560-17 [ Links ]
82. Lam AM, Ren S, Espiritu C, et al. Hepatitis B virus capsid assembly modulators, but not nucleoside analogs, inhibit the production of extracellular pregenomic RNA and spliced RNA variants. Antimicrob Agents Chemother 2017;61(8):e00680. https://doi.org/10.1128/AAC.00680-17 [ Links ]
83. Lee AC. Exploring combination therapy for curing HBV: Preclinical studies with capsid inhibitor AB-423 and a siRNA Agent, ARB-1740. Hepatology 2016; 63(1 Suppl):122A. [ Links ]
84. Berke JM, Dehertogh P, Vergauwen K, et al. Capsid assembly modulators have a dual mechanism of action in primary human hepatocytes infected with hepatitis B virus. Antimicrob Agents Chemother 201725;61(8):e00560-17. https://doi.org/10.1128/AAC.00560-17 [ Links ]
85. Vandyck K, Rombouts G, Stoops B, et al. Synthesis andevaluation of N-phenyl-3-sulfamoyl-benzamide derivatives as capsid assembly modulators inhibiting hepatitis B virus (HBV). J Med Chem 2018. ePub ahead of print. https://doi.org/10.1021/acs.jmedchem.8b00654 [ Links ]
86. Ding Y, Zhang H, Niu J, et al. PS-046-Multiple dose study of GLS4JHS, interfering with the assembly of hepatitis B virus core particles, in patients infected with hepatitis B virus. J Hepatol 2017;66(1):S27-S28. [ Links ]
87. Liu J, Zhang E, Ma Z, et al Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD-L1 blockade in chronic hepadnaviral infection. PLoS Pathog 2014;10(1):e1003856. https://doi.org/10.1371/journal.ppat.1003856 [ Links ]
88. Lanford RE, Guerra B, Chavez D, et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013;144(7):1508-1517. https://doi.org/10.1053/j.gastro.2013.02.003 [ Links ]
89. Gane EJ, Lim YS, Gordon SC, et al. The oral toll-like receptor-7 agonist GS-9620 in patients with chronic hepatitis B virus infection. J Hepatol 2015;63(2):320-328. https://doi.org/10.1016/j.jhep.2015.02.037 [ Links ]
90. Pallett LJ, Gill US, Quaglia A, et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat Med 2015;21(6):591-600. https://doi.org/10.1038/nm.3856 [ Links ]
91. King TH, Kemmler CB, Guo Z, et al. A whole recombinant yeast-based therapeutic vaccine elicits HBV X, S and core specific T cells in mice and activates human T cells recognizing epitopes linked to viral clearance. PLoS One 2014;9(7):e101904. https://doi.org/10.1371/journal.pone.0101904 [ Links ]
92. Hu J, Liu K. Complete and incomplete hepatitis b virus particles: Formation, function, and application. Viruses 2017;9(3):e56. https://doi.org/10.3390/v9030056 [ Links ]
93. Liu J, Yang HI, Lee MH, et al. Spontaneous seroclearance of hepatitis B seromarkers and subsequent risk of hepatocellular carcinoma. Gut 2014; 63(10):1648-1657. https://doi.org/10.1136/gutjnl-2013-305785 [ Links ]
94. Lauret E, Gonzalez-Dieguez ML, Rodriguez M, et al. Long-term outcome in Caucasian patients with chronic hepatitis B virus infection after HBsAg seroclearance. Liver Int 2015;35(1):140-147. https://doi.org/10.1111/liv.12461 [ Links ]
95. Lok AS, Zoulim F, Dusheiko G, Ghany MG. Hepatitis B cure: From discovery to regulatory approval. J Hepatol 2017;67(4):847-861. https://doi.org/10.1016/j.jhep.2017.05.008 [ Links ]
 Correspondence:
Correspondence:
G Dusheiko
(g.dusheiko@ucl.ac.uk


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}