










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.108 n.4 Pretoria Apr. 2018
http://dx.doi.org/10.7196/samj.2018.v108i4.12823
RESEARCH
Hereditary angio-oedema in the Western Cape Province, South Africa
K M CoovadiaI; M-Y ChothiaII; S G BakerIII; J G PeterIV, V; P C PotterVI, VII
IMB ChB, FCP (SA), MMed, Dip Allerg (SA); Department of Internal Medicine, Tygerberg Hospital and Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
IIMB ChB, FCP (SA), MMed, Cert Nephrology (SA); Department of Internal Medicine, Tygerberg Hospital and Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
IIIBSc (Nursing), MSc (Medicine), Dip Asthma (UK); Allergy Diagnostic and Clinical Research Unit, University of Cape Town Lung Institute, Cape Town, South Africa
IVMB ChB, MMed, FCP (SA), PhD; Allergy Diagnostic and Clinical Research Unit, University of Cape Town Lung Institute, Cape Town, South Africa
VDivision of Allergology and Clinical Immunology, Department of Internal Medicine, Groote Schuur Hospital and Faculty of Health Sciences, University of Cape Town, South Africa
VIMB ChB, DCH (SA), FCP (SA), BSc Hons Immunology, MD, FAAAAI, FACAAI; Allergy Diagnostic and Clinical Research Unit, University of Cape Town Lung Institute, Cape Town, South Africa
VIIMB ChB, DCH (SA), FCP (SA), BSc Hons Immunology, MD, FAAAAI, FACAAI; Division of Allergology and Clinical Immunology, Department of Internal Medicine, Groote Schuur Hospital and Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
BACKGROUND. Hereditary angio-oedema (HAE) is an autosomal dominant condition caused by a deficiency in the C1-esterase inhibitor protein, resulting in increased bradykinin release. It presents clinically with recurrent attacks of angio-oedema, commonly affecting the limbs, face, upper airway and gastrointestinal tract. Little is known about this condition in sub-Saharan Africa.
OBJECTIVES. To analyse and report on the clinical presentation and treatment of patients with HAE in the Western Cape Province, South Africa.
METHODS. A retrospective analysis was conducted on a series of 60 cases of HAE seen between 2010 and 2015 at the Allergy Diagnostic and Clinical Research Unit, University of Cape Town Lung Institute, and the Allergy Clinic at Groote Schuur Hospital, Cape Town. The findings in 43 cases of type 1 HAE are described. Parameters assessed included age, gender, age of diagnosis, duration of illness, family history, identifiable triggers, average duration of attack, number of attacks per year and type of attack.
RESULTS. A total of 43 patients were included in this study. Of these, 65.1% (28/43) were female. The median age at diagnosis was 20 years (interquartile range (IQR) 10 - 27) and the median duration of illness 10.5 years (IQR 6 - 22). Of the patients, 62.8% (27/43), 32.6% (14/43) and 4.7% (2/43) were of mixed ancestry, white and black African, respectively; 51.2% (22/43) were index cases, with the remaining 48.8% (21/43) diagnoses via family member screening, 12 families making up the majority of the cohort. The mean (standard deviation) duration of an acute attack was 49 (25.8) hours, and 64.3% (27/42), 71.4% (30/42), 14.3% (6/42) and 88.1% (37/42) of patients experienced facial or upper airway, abdominal, external genitalia and limb attacks, respectively. Danazol for long-term prophylaxis was used in 21 patients, while C1-inhibitor concentrate (Berinert) was accessed for short-term prophylaxis in only four patients. Acute life-threating attacks were treated with fresh frozen plasma in 11 patients, and only four accessed icatibant. The mortality rate for the period 2010 - 2015 was 4.5% (2/43). The prevalence of HAE in the Western Cape was estimated to be 1:140 000.
CONCLUSIONS. HAE occurs in South Africans of all ethnicities, and life-threatening attacks occur in almost two-thirds of patients. Despite limited therapeutic options and very limited access to gold-standard therapies available in the developed world, our mortality rate is very low, with both the deaths related to inability to access emergency treatment rapidly.
Hereditary angio-oedema (HAE) is a life-threatening autosomal dominant condition caused by a deficiency in the C1-esterase inhibitor (C1-INH), a pivotal protein in the complement and fibrinolytic pathways.[1] The condition was first described over 100 years ago,[2] yet little is known about it in sub-Saharan Africa, with only an isolated report of a case in a Zulu kindred having appeared in the literature.[3]
Over a period of 35 years, 60 cases of HAE have been identified between the Allergy Clinic at Groote Schuur Hospital, Cape Town, South Africa (SA), and the Allergy Diagnostic and Clinical Research Unit (ADCRU) at the University of Cape Town (UCT) Lung Institute. To our knowledge, this is the largest cohort of HAE patients on the African continent. This study describes a series of adult patients with HAE living in the Western Cape Province.
Objectives
To retrospectively analyse the adult cases of type 1 HAE identified in the Western Cape cohort.
Methods
We reviewed the clinical records of all cases of HAE that were diagnosed at ADCRU and the Groote Schuur Hospital Allergy Clinic. A total of 60 patient records were reviewed. After excluding 17 cases (7 patients did not live in the Western Cape, and the rest were minors (aged <18 years) diagnosed as index cases), 43 cases were included in our analysis. In all these 43 patients, type 1 HAE was confirmed by laboratory tests including functional C1-INH activity assays when available and/or determination of antigenic levels of the C1-INH enzymes to patient serum. C4 levels were measured in each patient. These patients either presented as index cases or were diagnosed after a family member had been diagnosed with type 1 HAE.
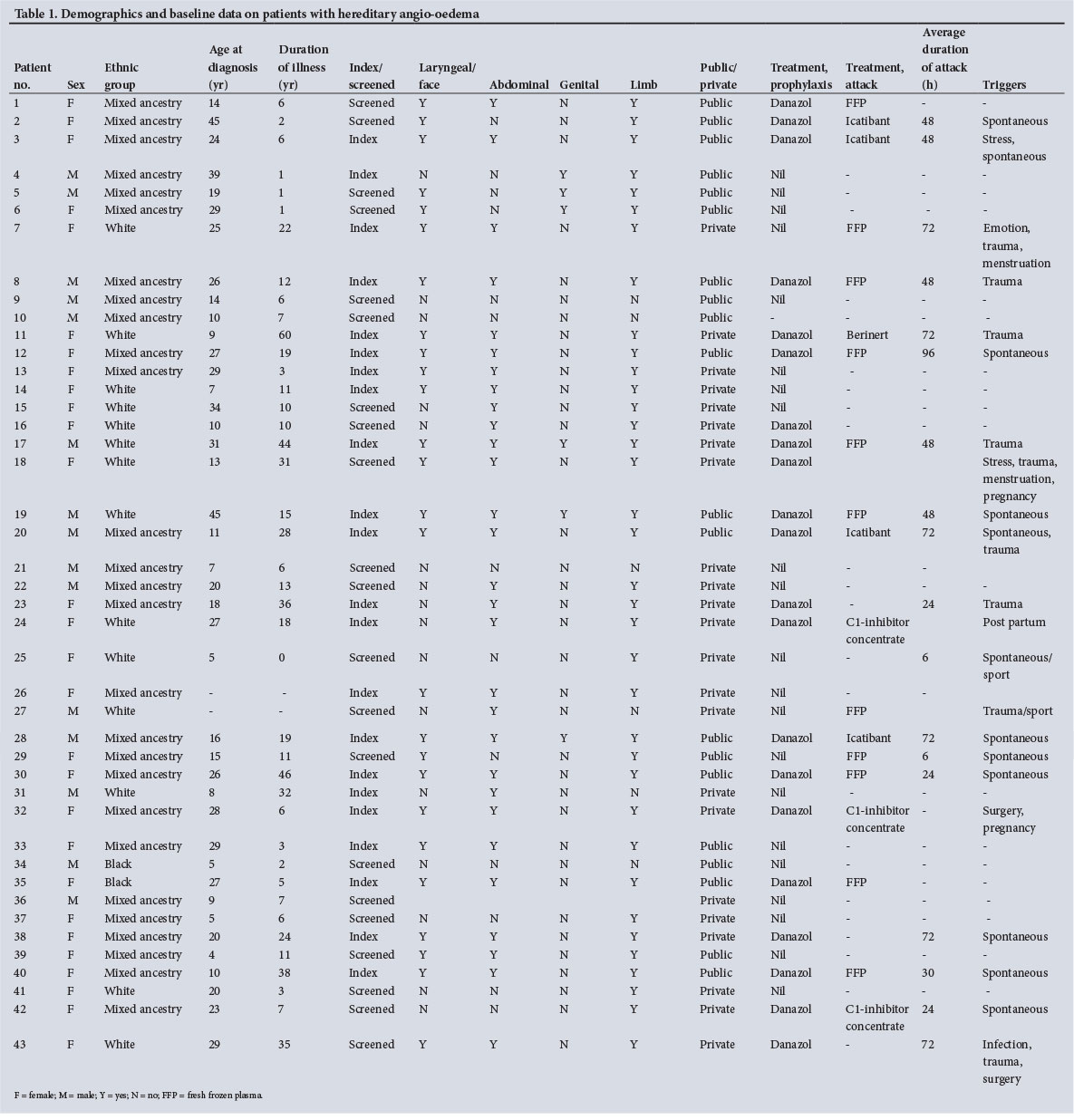
The parameters reviewed were age, gender, age of diagnosis, duration of illness, distribution of angio-oedema, diagnostic tests, family history, identifiable triggers, average duration of attack, number of attacks per year and type of attack (Table 1). Treatment regimens utilised for each patient were also reviewed.
Ethical considerations
Ethics approval was obtained from the Stellenbosch University Human Research Ethics Committee (ref. no. S16/05/090) and the UCT Human Research Ethics Committee (ref. no. 462/2016). The study was conducted in accordance with the ethical principles set out in the Declaration of Helsinki (2013), and permission to study the patients was obtained from the medical superintendent of Groote Schuur and Tygerberg hospitals.
Patient confidentiality was maintained at all times by allocating a study number to each patient prior to removing identifiable personal information from patients' files and collating them in a data collection sheet.
Results
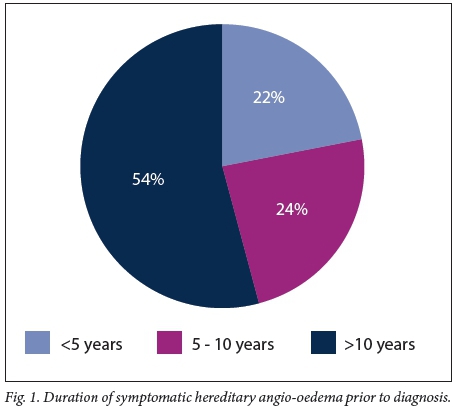
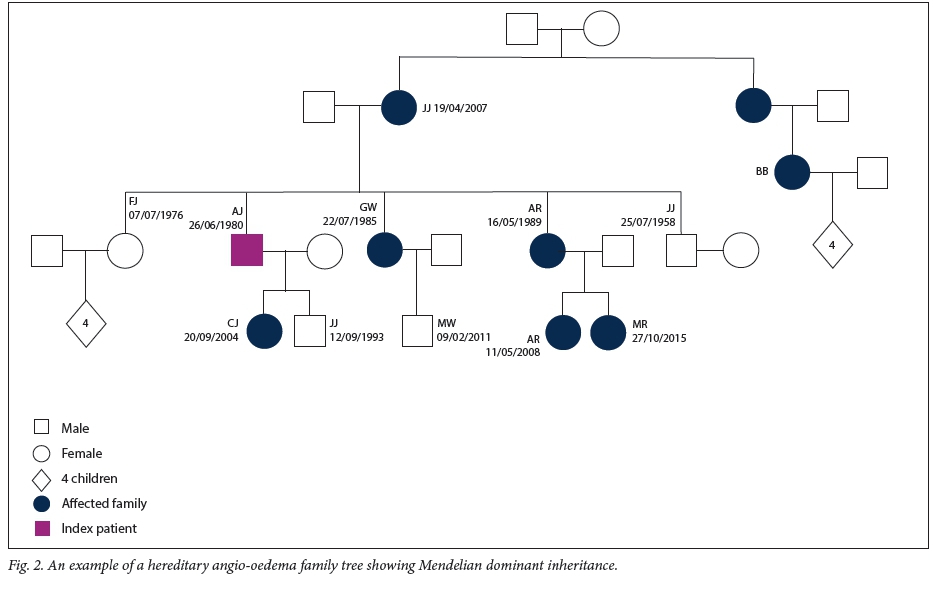
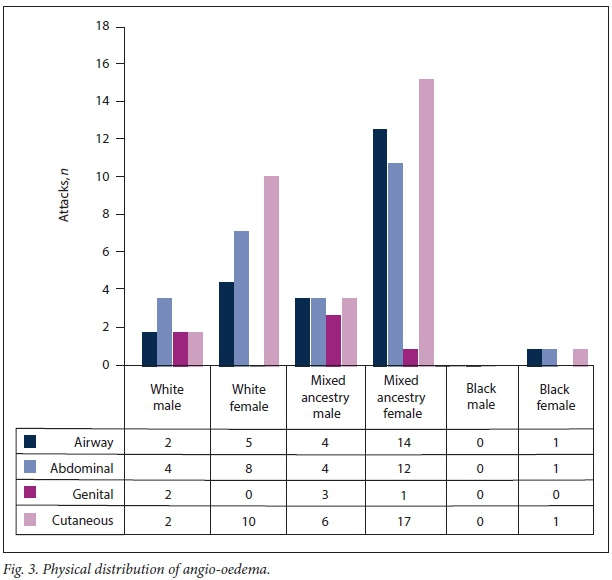
Baseline data for the 43 patients reported in this study are set out in Table 1. Of the patients, 65.1% (28/43) were female. The median age at diagnosis was 20 years (interquartile range (IQR) 10 - 27) and the median duration of illness under care 10.5 years (IQR 6 - 22) (n=42) (Fig. 1). Of the patient cohort, 62.8% (27/43) were of mixed ancestry, 32.6% (14/43) white and 4.7% (2/43) black African; patient ethnicities were self-reported during screening. The majority of the patients were index cases (51.2%, 22/43), with the remaining 48.8% (21/43) diagnosed via family member screening. Overall, 12 families were screened for HAE in this cohort. No patient with type 2 HAE was identified. An example of a family tree showing Mendelian dominant inheritance is provided in Fig. 2. The mean (standard deviation) duration of an acute attack was 49 (25.8) hours. The only missing basic clinical data were 2 patients without age at diagnosis, 2 without duration of illness, 1 without anatomical distribution of oedema, 3 without C4 levels and 4 without C1-INH levels; however, only 18 patients had data on the duration of acute attacks. The mortality rate for the period 2010 - 2015 was 4.5% (2/43). Just over half (53.0%, 23/43) of our patients were seen as private patients, the remaining 46.5% (20/43) being treated in the public healthcare sector. The distribution of attacks is shown in Fig. 3. There were no significant differences when analysing by ethnicity or gender.
Danazol for long-term prophylaxis was used in 48.8% of patients (21/43). Most patients were treated with doses between 100 and 200 mg/d without significant side-effects (hypertension, dyslipidaemia, hirsutism, virilisation and hepatic disease) being detected at their 6-monthly follow-up visits. Liver function tests were done at each follow-up visit. Only four patients (9.3%) used C1-inhibitor concentrate (Berinert) as short-term prophylaxis during elective surgery. The majority (66.0%) were well controlled on danazol only. A total of 19 patients (44.2%) were treated for life-threatening attacks involving the airway or gastrointestinal tract. Two of these patients had stopped long-term danazol prophylaxis prior to their planned pregnancies. One patient received C1-inhibitor concentrate during an acute attack involving the upper airway, and responded rapidly with relief of symptoms after the infusion. Fresh frozen plasma (FFP) was used for acute attacks in 25.6% of patients (11/43), 8 in the public sector and 3 in the private sector. Icatibant was used to treat 9.3% of patients (4/43) for life-threatening attacks. Patients with laryngeal or abdominal attacks required admission to an intensive care unit (ICU) for treatment and observation for 3 - 5 days. These patients had an average of 11.75 life-threatening attacks per annum, despite being on maximal doses of danazol prophylaxis. The group treated with icatibant demonstrated rapid resolution of symptoms during an attack, with symptom relief starting as early as 20 minutes post injection and full resolution within 4 hours in the majority of cases. No adult patients in our case series were treated with tranexamic acid.
Most patients reported spontaneous onset of attacks, but others identified triggers that included minor limb trauma (e.g. tight shoes, gardening), emotional stress, menstruation, pregnancy and surgery.
The mortality rate for the period 2010 - 2015 was 4.7% (2/43).
These two patients died as a result of laryngeal asphyxia. Both lived far from a hospital and died as a result of delayed access to a high-care medical facility associated with transport delays. Another patient was admitted to an ICU and effectively treated with FFP four times during the study period for recurrent laryngeal angio-oedema, but did not require a tracheostomy.
Discussion
In 1888, Sir William Osler[2] was the first to describe a case of HAE, in a 24-year-old woman who presented with a lifelong history of recurrent episodes of painless, non-pruritic swellings of her body, each lasting between 1 and 4 days. These swellings affected various parts of her body - hands, fingers, kneecaps, elbows, face and lips -while some severe episodes presented with abdominal colic, nausea and vomiting. The attacks occurred spontaneously while she was feeling well, with no specific triggers identified. She reported that her mother suffered from similar symptoms. Osler then obtained a comprehensive family history, which revealed that these symptoms had been present in 28 family members in the preceding five generations, and noted that the patients experienced local swellings in various parts of the body - face, hands, arms, legs, genitals, buttocks and throat. In one instance, death resulted from sudden oedema glottidis. The oedema was almost invariably associated with gastrointestinal disturbance, patients experiencing colic, nausea, vomiting and sometimes diarrhoea. Furthermore there was a marked hereditary disposition, the disease having affected members of the family in five generations.
It was not until 1963 that Donaldson and Evans[4] used immunoelectrophoresis to demonstrate in their own series of patients, with similar symptoms to those described by Osler, a deficiency of C1-INH. We know today that HAE is a rare autosomal dominant disorder caused by mutations of the C1-INH gene, resulting in a quantitative or qualitative deficiency of C1-INH. Over 280 different C1-INH gene mutations have been identified so far, with a spontaneous mutation rate of 25%.[5,6]
HAE manifests clinically as recurrent episodes of non-pruritic oedema that can affect any part of the body, commonly involving structures of the upper airways, gastrointestinal tract, external genitalia and extremities, with up to a quarter of patients reporting local erythema prior to the onset of swelling. HAE can be distinguished from other forms of angio-oedema (IgE-mediated and physical forms) because attacks usually last longer (72 - 96 hours) and are not associated with pruritus and urticaria. Symptoms are not alleviated by steroids or antihistamines.[7] Central to the pathogenesis of type 1 HAE is a deficiency of C1-INH. This is a serine protease inhibitor belonging to the SERPING-1 family that serves as a major inhibitor of the complement system, proteases, kallikrein and Hageman factor (coagulation factor XIIa). Along with these functions, it also inhibits the formation of bradykinin from kininogen. A deficiency of C1-INH therefore causes an increase in kallikrein activity, resulting in excessive production of bradykinin.[5,6]
Bradykinin has been demonstrated to be the primary mediator responsible for the development of angio-oedema. It binds to the bradykinin β2 receptor on vascular endothelial cells, resulting in significant increases in vascular permeability. This increase in vascular permeability is thought to be due to its effect on endothelial tight junctions responsible for the regulation of water movement across the endothelial membrane.[6]
Worldwide incidences of HAE have been reported to range from 1:10 000 to 1:150 000. [8] There is no gender or racial predominance for type 1 or type 2 HAE, but most reported cases of type 3 HAE have been in females.[9] Type 1 HAE accounts for 80% of cases globally. Type 2 and type 3 HAE are rare conditions. In ~80% of reported cases of HAE-1 there is a positive family history, while 20% of new cases are attributed to de novo mutations of the SERPING1 protein.[9] There is considerable variation in interpersonal and intrapersonal severity of attacks, even among family members with the same gene defect. Most patients experience their first attack by the age of 15 years, with the mean age of onset of symptoms being 8 - 12 years.[10]
Typically the onset of oedema is described as crescendo in nature, developing over several hours, peaking at 12 - 24 hours and then resolving over the next 72 - 96 hours.[11] Commonly identified triggers range from surgical procedures (dental procedures being the most common) to pregnancy, menstruation, minor trauma (sports, gardening, tight shoes), emotional stress, sepsis and drugs (oestrogen therapy and angiotensin-converting enzyme inhibi-tors).[11,12] Abdominal symptoms are dominant in up to 25% of cases and manifest as intestinal colic due to intestinal wall and mesenteric oedema. Patients typically present with abdominal pain, nausea and vomiting.[11]
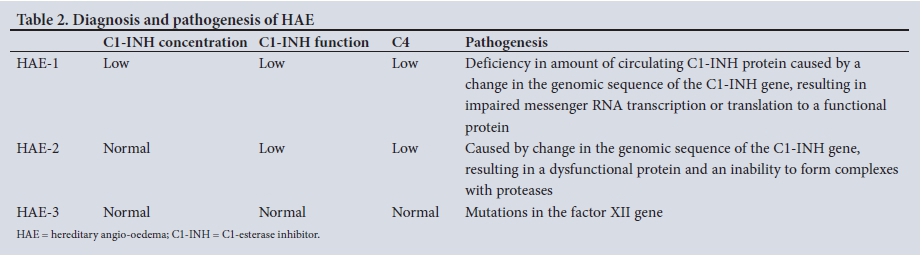
Life-threatening angio-oedema is the result of asphyxia due to swelling of the upper airway structures, usually the larynx, which accounts for the 15 - 30% mortality rate.[11] C4 levels are a useful screening test (especially in primary healthcare facilities), as low serum C4 levels are a consistent feature of untreated HAE. Patients who present with clinical features of angio-oedema and have normal levels of circulating C4 are unlikely to have HAE. The diagnosis of type 1 HAE is made by confirming low levels of antigenic C1-INH protein. However, if C1 esterase levels are normal or elevated (along with a low C4 level), functional C1-INH testing should be conducted to confirm type 2 HAE[6,8,11,12] (Table 2). Based on the data presented, the estimated prevalence of type 1 HAE in adults living in the Western Cape is 1:140 000. Possible explanations for the apparent low prevalence may be underdiagnosis of cases by attending doctors, or lack of referral of rural patients to a tertiary centre for further evaluation. A recently published study from Italy showed a prevalence of 1:64 935.[13] The median age of diagnosis in our case series was 20 years (IQR 10 - 27), while that in Italy was 25 years;[13] 67.4% (29/43) of our patients were female, which is not significantly different from the 53% female prevalence in the Italian study.[13] Most (60.5%, 26/43) of our patients were diagnosed as index cases, with no evident family history. The reported incidence of de novo mutations is 25%.[6]
Female patients with HAE are often more symptomatic than male patients. Oestrogens have been implicated in worsening the course of the disease[14] and make treatment of female HAE patients challenging with regard to oral contraception, pregnancy, surgical procedures and menopause. The side-effects of long-term use of attenuated androgens (hirsutism, virilisation, hypertension and mood changes) negatively affect female patients more than males, making compliance more challenging. Androgens are contraindicated for use during pregnancy, especially in the first trimester. Pregnancy has a variable effect on the severity and frequency of attacks.
Very few patients in SA have access to plasma-derived human Cl-inhibitor concentrate (Berinert), which is the globally recommended form of prophylaxis for females during pregnancy.[7] The microsomal cytochrome p450 enzyme aromatase can convert androgens to oestrogens.[l4] Danazol is therefore not recommended for use in patients who suffer from both HAE and breast cancer. There has been no documented decrease in fertility in females taking attenuated androgens for prophylaxis, but impaired fertility has been reported in males on this treatment. [14]
The standard prophylactic therapy for patients with HAE is danazol, an attenuated androgen. Prophylaxis with danazol is stopped in our patients who become pregnant (Table 3). One of our patients received a prophylactic Cl-inhibitor concentrate intraoperatively for a caesarean section and reported no severe attacks during the peripartum period. She did, however, experience an increase in frequency of attacks during her pregnancy, while she was off danazol prophylaxis. Some of our patients have used Cl-inhibitor concentrate as preoperative prophylaxis for major surgical procedures (e.g. hip replacement surgery).
HAE poses many challenges in SA. The major difficulty lies in not recognising the condition and its various clinical manifestations. Those who correctly clinically diagnose HAE are further challenged in the diagnostic work-up and treatment of these patients. Currently only C4 levels and Cl-INH quantitative testing are available in SA through the National Health Laboratory Service and a few private laboratories. Functional Cl-INH assays are not currently available for clinicians, and would be important in the diagnosis of type 2 HAE. We have not yet identified any patients with type 2 HAE. Functional assays can, however, be sent to European laboratories for evaluation. Serum specimens have to be shipped frozen, as the enzyme is heat labile. This is expensive and logistically difficult.
Access to novel therapies such as icatibant and Cl-inhibitor concentrate, which are widely available in Europe and North America, are generally limited to only a few individuals in SA who have adequate medical insurance schemes. However, a state hospital has recently approved the use of Cl-inhibitor concentrate for a young pregnant adult HAE patient. These medications are not yet registered in SA and have to be individually imported after obtaining permission via the Medicines Control Council (MCC). Furthermore, the cost of such medication has made it unaffordable for many patients. Most patients in the public health sector do not yet have access to these lifesaving treatments, recommended in global guidelines.[7]
In developed countries, 'on-demand' therapy,'[5] whereby patients self-treat at home on the onset of symptoms and swellings, is available. SA patients, however, are dependent on clinicians initiating therapy with FFP once the disease has progressed. FFP is only given for abdominal or life-threatening upper airway attacks. However, there is limited documentation on the efficacy of FFP in the management of life-threatening angio-oedema in the literature.'" This extended duration of treatment often results in longer recovery periods, more days off work and an overall negative economic impact. A 3-day admission to an ICU costs ~ZAR58 000, while an 'on demand' alternative, icatibant or Cl-inhibitor concentrate, currently costs ~ZARl7 000 per treatment - with the added benefit of convenience and possibly enabling the patient to return to work the same day. Cl-inhibitor concentrate can be used for acute attacks or for prophylaxis. It is safe and is also recommended for use in the paediatric age group.'[7]
Many patients experience minor attacks involving the limbs. These are often self-limiting and the patient may not need FFP or expensive treatments such as Cl-inhibitor concentrate or icatibant for every attack.
'On-demand' therapy with the selective bradykinin-ß2-receptor antagonist icatibant is the gold standard of therapy for acute attacks in many developed countries. Patients are taught to recognise early symptoms of an attack and inject 30 mg subcutaneously, as published in both the FAST-l and FAST-2 trials.'[8]
Most patients respond well to the usual dose of danazol prophylaxis (200 mg/d). Some patients in our cohort were adequately controlled on l00 mg/d. Patients should be managed with the lowest possible dose that controls symptoms adequately. While 80.0% of the patients in our cohort observed over a l-year period were well controlled on danazol prophylaxis alone, the remaining 20% were noted to have recurrent or severe attacks. Despite being on maximal doses of danazol, patients in this subgroup experienced an average of ll.5 life-threatening attacks a year. Patients who have occasional minor attacks three or four times a year may be monitored without danazol prophylaxis. This is often the case with adolescent patients, who experience a few minor attacks per year.
Four patients in our cohort were treated with icatibant owing to their increased frequency of life-threatening attacks despite being compliant on maximal doses of danazol. Treatment with icatibant not only improved symptom alleviation (most reported improvement by 20 minutes post injection, and full resolution by 4 hours) but also decreased the financial burden of lost work days. Prior to treatment with icatibant, some of these patients had reported an estimated attack duration of 48 - 72 hours. Icatibant is also not yet licensed for use in SA unless imported on a named-patient basis (with approval from the MCC). On-demand therapy should be made available to all South Africans with HAE, as it can be self-administered at home and does not require the patient to present to a healthcare facility. Icatibant is only indicated for acute attacks, and not for prophylaxis prior to undergoing surgical procedures.
While paediatric patients (aged <13 years) were not included for reporting in this study, they made up a significant proportion of our total cohort (10/60). Their cases will be the subject of a separate report. These patients are managed by paediatricians if they present as index cases, or are being closely monitored if they are diagnosed in the course of screening and remain asymptomatic. Prophylaxis is the cornerstone of preventive management in asymptomatic adult patients, but is not usually recommended in asymptomatic paediatric patients. Children can be treated with tranexamic acid (on a per kilogram dosage), but some children require danazol at low doses (per kilogram) to prevent recurrent attacks. C1-inhibitor concentrate is safe for use in children and should be made available to all age groups. Adolescent patients are managed in the adult clinics and are treated like adult cases.
In addition to increasing physician awareness of HAE, it would be beneficial if SA patients had support groups available locally. Some of our patients belong to international support groups (e.g. HAEi, the international umbrella organisation for the world's HAE patient groups), but face challenges very different to their overseas counterparts, in particular access to the first-line treatments recommended in the International Global Consensus Guidelines for HAE.[7] Our challenges include obtaining approval for novel therapies via medical insurance schemes, arranging for emergency transportation to appropriate emergency rooms, and education of the medical profession and medical aid schemes on the efficacy of the new life-saving treatments. Transportation can pose a huge challenge in SA. One of our patients died as a result of laryngeal asphyxia while wating for an ambulance. Local HAE support groups will assist in making living with HAE in SA more manageable and in helping to improve doctor and public awareness.
All patients with a diagnosis of HAE should wear a Medic-Alert bracelet and have a written action plan for the treatment of acute attacks. They should also be notified to the South African Primary Immunodeficiency Register after providing the necessary consent.
Conclusion
This is the first large series of HAE to be reported on the African continent. HAE, rare in SA, is a life-threatening condition. Every clinician, particularly those working in emergency units, should have a standardised approach to angio-oedema and its management. HAE is not an IgE-mediated form of angio-oedema and therefore will not respond to corticosteroid or adrenaline therapy. While current practice in SA is in keeping with global guidelines for maintenance prophylaxis, treatment of HAE with novel therapies for acute attacks is limited to only a few select patients. With increased physician awareness, more patients with HAE will be correctly diagnosed. This will encourage global pharmaceutical companies to register novel therapies in SA. SA-based patient support groups are needed to assist patients living with HAE in this country and adequately address our unique challenges.
Acknowledgements. We thank Ms Kathryn Manning (statistician), Ms Lindi Foot, the staff at ADCRU and the Groote Schuur Hospital Allergy Clinic, and Dr F M Coovadia.
Author contributions. Equally contributing authors.
Funding. None.
Conflicts of interest. None.
References
1. Frank MM. Hereditary angioedema: A current state-of-the-art review, VI: novel therapies for hereditary angioedema. Ann Allergy Asthma Immunol 2008;100(Suppl 2):S23-S29. https://doi.org/10.1016/S1081-1206(10)60583-2 [ Links ]
2. Osler W. Hereditary angio-neurotic oedema. Am J Med Sci 1888;95(4):362-367. [ Links ]
3. Moran E, Isaacs GS, Naidoo B, et al. Hereditary C1 esterase deficiency in a Zulu kindred. S Afr Med J 2009;99(1):40-42 [ Links ]
4. Donaldson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: Absence of serum inhibitor of C1-esterase. Am J Med 1963;35(1):37-44. [ Links ]
5. Kaplan A, Joseph K. The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol 2010;104(3):193-204. https://doi.org/10.1016/j.anai.2010.01.007 [ Links ]
6. Zuraw BL. The pathophysiology of hereditary angioedema. World Allergy Organ J 2010;3(Suppl 3):S25-S28. https://doi.org/10.1097/1939-4551-3-S3-S25 [ Links ]
7. Lang DM, Aberer W, Bernstein JA, et al. International consensus on hereditary and acquired angioedema. Ann Allergy Asthma Immunol 2012;109(6):395-402. https://doi.org/10.1016/j.anai.2012.10.008 [ Links ]
8. Zuraw BL. Clinical practice: Hereditary angioedema. N Engl J Med 2008;359(10):1027-1036. https://doi.org/10.1056/NEJMcp0803977 [ Links ]
9. Craig T, Riedl M, Dykewicz MS, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol 2009;102(5):366-372. https://doi.org/10.1016/S1081-1206(10)60506-6 [ Links ]
10. Bork K, Meng G, Staubach P, et al. Hereditary angioedema: New findings concerning symptoms, affected organs, and course. Am J Med 2006;119(3):296-274. https://doi.org/10.1016/j.amjmed.2005.09.064 [ Links ]
11. Gompels MM, Lock RJ, Abinun M, et al. C1 inhibitor deficiency: Consensus document. Clin Exp Immunol 2005;139(3):379-394. https://doi.org/10.1111/j.1365-2249.2005.02726.x [ Links ]
12. Gompels MM, Lock RJ, Morrison L, et al. Primary immunodeficiency association: Consensus document for the diagnosis and management ofC1 inhibitor deficiency. Clin Exp Immunol 2005;139(3):379-394. https://doi.org/10.1111/j.1365-2249.2005.02726.x [ Links ]
13. Zanichelli A, Arcoleo F, Barca MP, et al. A nationwide survey of hereditary angioedema due to C1 inhibitor deficiency in Italy. Orphanet J Rare Dis 2015;10:11. https://doi.org/10.1186/s13023-015-0233-x [ Links ]
14. Caballero T, Farkas H, Bouillet L, et al. International consensus and practical guidelines on gynaecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol 2012; 129(2):308-320. https://doi.org/10.1016/j.jaci.2011.11.025 [ Links ]
15. Aygoren-Pursun E, Saguer IM, Rusicke E, et al. On demand treatment and home therapy of hereditary angioedema in Germany - the Frankfurt experience. Allergy Asthma Clin Immunol 2010;6:21. https://doi.org/10.1186/1710-1492-6-21 [ Links ]
16. Tang R, Chen S, Zhang HY. Fresh frozen plasma for the treatment of hereditary angioedema acute attacks. Chin Med Sci J 2012;27(2):92-95. [ Links ]
17. Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol 2009;124(4):801-808. https://doi.org/10.1016/j.jaci.2009.07.017 [ Links ]
18. Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med 2010;363(6):532-541. https://doi.org/10.1056/NEJMoa0906393 [ Links ]
 Correspondence:
Correspondence:
KM Coovadia
kcoovadia@gmail.com
J G Peter
jonny.peter@uct.ac.za
Accepted 26 October 2017


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}