










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.108 n.3 Pretoria Mar. 2018
http://dx.doi.org/10.7196/samj.2018.v108i3.13158
CME
N AlliI; J VaughanII; S LouwIII; S MoodlyIV; M PatelV
IMB BCh, FCPathHaem (SA);Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
IIMB BCh, FCPathHaem (SA), MMed Haem; Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
IIIMB BCh, FCPathHaem (SA), MMed Haem; Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
IVNDip MedTech, NHD MedTech, MSc Medicine; Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
VMB ChB, FCP (SA), MMed, FRCP (Lond), PhD; Department of Clinical Haematology, Division of Internal Medicine, Chris Hani Baragwanath Academic Hospital, Johannesburg, and School of Medicine, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Bleeding disorders are divided into two broad categories, i.e. inherited, discussed in part 1 of this CME series, and acquired, which is the subject of discussion in the current issue. In contrast to inherited haemorrhagic disorders, where generally a single haemostatic abnormality is found, multiple haemostatic defects are commonly present in acquired haemorrhagic diseases. Bleeding is often a presenting manifestation of systemic disease and requires a multidisciplinary team approach. Iatrogenic causes of abnormal haemostasis are of particular importance to the emergency physician. This CME article aims to provide an approach to the diagnosis and management of acquired bleeding disorders encountered in general practice.
Acquired bleeding disorders encompass a heterogeneous group of conditions with varied and often complex aetiologies. Clinical evaluation of patients presenting with a bleeding disorder often provides clues as to whether the abnormality resides in coagulation factors, platelets or blood vessels. A detailed history and complete physical examination are therefore imperative for meaningful interpretation of laboratory tests, as complex haemostatic derangements may accompany specific clinical scenarios. Interpretation based solely on laboratory tests may be misleading.
For discussion purposes, acquired bleeding disorders are divided into the following groups: (i) clotting factor deficiencies; (ii) abnormalities of platelet number or function; (iii) vascular defects; or (iv) various combinations of the three abovementioned disorders. The last group includes liver disease, disseminated intra-vascular coagulation (DIC) and chronic kidney disease.
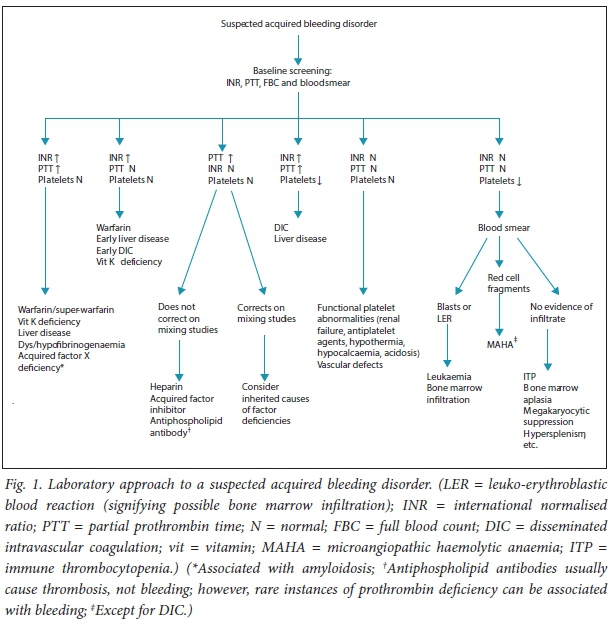
Basic laboratory tests (partial thrombo-plastin time (PTT), international normalised ratio (INR), full blood count (with peripheral blood smear assessment)) and accurate clinical information form a basis for further investigations. To this end, an algorithmic approach is incorporated to serve as a guide (Fig. 1).
Clotting factor deficiencies
Coagulation factor inhibitors
Coagulation factor inhibitors are antibodies that neutralise specific coagulation factors. These antibodies can develop against any factor in the coagulation cascade, but factor VIII (FVIII) is most frequently involved, and may develop in patients with inherited haemophilia A as an immune response to factor-replacement therapy, or spontaneously as auto-antibodies, resulting in the bleeding condition termed acquired haemophilia. The presence of inhibitors is suspected in a patient with abnormal bleeding without any prior bleeding diathesis, or when a patient with known haemophilia has more extreme bleeding than usual or fails to achieve haemostasis after factor replacement. Prolongation of the screening clotting assays, i.e. INR and/or PTT, with failure to correct on mixing with normal plasma, should alert the attending clinician to the presence of an inhibitor.
Factor inhibitors in inherited haemophilia A and B
This has been discussed in part 1 of this series.[1]
Acquired haemophilia A
Acquired haemophilia A (AHA) is a rare (~1 per million of the population per year), potentially life-threatening auto-immune bleeding disorder due to inhibiting auto-antibodies (inhibitors) against endogenous FVIII. Although documented as a rare condition, AHA is probably under-diagnosed. Mortality in cases of AHA exceeds 20% in patients >65 years of age and those with comorbid disease, such as underlying malignancies. Although death directly due to excessive bleeding is not common, it does contribute to morbidity, thereby increasing the duration and cost of hospitalisation (e.g. delayed wound healing and increased transfusion requirements). Immune modulation therapy to eradicate the inhibitor also contributes significantly to cost and mortality. In contrast to inherited haemophilia, AHA affects both males and females and is most common in the elderly (median age 64 - 78 years). AHA can, however, occur in younger patients in relation to pregnancy and auto-immune diseases. Most cases are idiopathic, but underlying precipitating causes include pregnancy, auto-immune diseases (most commonly rheumatoid arthritis), infection, malignancy and drugs (e.g. interferon alpha). Research suggests that the breakdown of immune tolerance to FVIII is due to both genetic and environmental factors. The majority (94.6%) of patients present with bleeding, which can either be spontaneous or provoked, e.g. after surgery. The site of bleeding is most commonly subcutaneous, followed by the gastrointestinal tract, intramuscular sites and genito-urinary tract. Bleeding in other sites (intracranial and retroperitoneal) can occur, but in contrast to congenital haemophilia, joint bleeding is infrequent. Because of the second-order (non-linear) kinetics of the anti-FVIII antibodies in AHA, FVIII levels are not predictive of bleeding risk and patients can have serious bleeding despite having only modestly reduced laboratory-determined FVIII activity levels.
Inhibitors to other coagulation factors
Inhibitors to other coagulation factors, i.e. fibrinogen, FII, FV, FVII, FIX, FX, FXI, FXIII and von Willebrand factor, do occur but are rare. Correct identification and quantification are, however, indicated for appropriate therapy. As with haemophilia A, these inhibitors develop either in patients with congenital deficiencies related to exposure to replacement therapy or spontaneously in people without a prior bleeding disorder. As with AHA, precipitating factors for spontaneous development of inhibitors include infections, drug exposure, auto-immune diseases, blood transfusions and underlying malignancies.[2]
Diagnosis
Depending on the position of the affected factor in the coagulation cascade, the screening assays, i.e. PTT and/or INR, will be prolonged. Other causes of prolonged screening tests, such as lupus anticoagulant and anticoagulant drugs, e.g. heparin, should be excluded prior to identification and quantification of the coagulation factor inhibitor.
The most common laboratory observation in AHA is a prolonged PTT that does not correct with mixing with normal plasma after incubation, together with a normal INR, thrombin time and platelet count.[2,3]
Management
Management of patients with acquired inhibitors entails: (i) control of bleeding with haemostatic agents, as for inherited haemophilia patients with inhibitors (part 1);[1](ii) eradication of the inhibitor with immune modulating agents (e.g. corticosteroids and rituximab); and (iii) treatment of the underlying pathogenic disease process. Thrombotic complications, including myocardial infarctions and cerebrovascular accidents, can occur in relation to haemostatic agent administration.[2-6]
Vitamin K deficiency
Vitamin K is responsible for gammacarboxylation of FII, FVII, FIX and FX, as well as for proteins C, S and Z. Gammacarboxylation enables binding to phospholipid membranes via Ca++ bridges. Vitamin K deficiency is encountered in various clinical scenarios and causes include: haemorrhagic disease of the newborn (currently termed vitamin K deficiency bleeding), reduced dietary intake, prolonged antibiotic use, cholestatic liver disease, malabsorption, and drugs, e.g. anticonvulsants and warfarin. The mode of therapy is oral or intravenous vitamin K, and patients with severe bleeding are treated with fresh-frozen plasma (FFP) or prothrombin complex concentrate (PCC).
Anticoagulation and antiplatelet agents Warfarin
Warfarin, a coumarin derivative, inhibits the enzyme vitamin K epoxide reductase and thereby impairs production of vitamin K-dependent coagulation factors, i.e. FII, FVII, FIX and FX, as well as proteins C, S and Z. Patients treated with coumarin derivatives have reduced concentrations of these coagulation factors, with consequent increased risk of bleeding that is amplified when the INR is supratherapeutic (particularly >5). Other factors that increase the bleeding risk include advanced age, a prior history of bleeding, previous stroke, hypertension, other drugs associated with a bleeding risk (such as non-steroidal anti-inflammatory drugs (NSAIDs)), and abnormal liver or renal function.
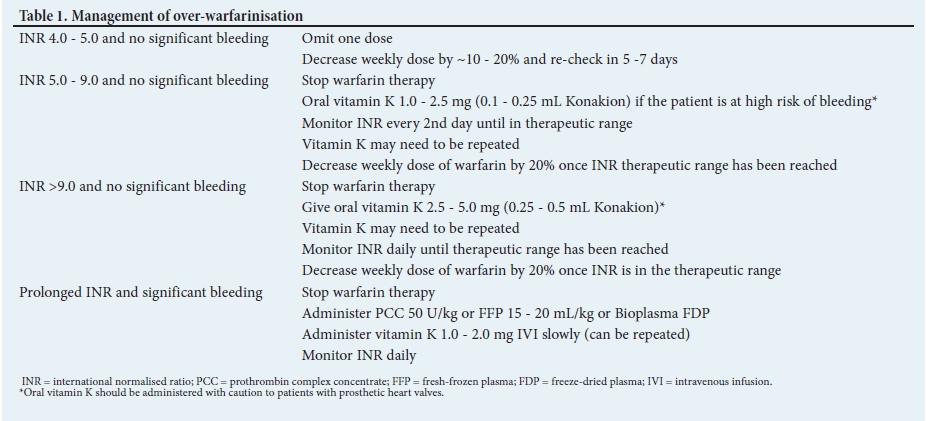
Management of warfarin-associated bleeding depends on the severity of bleeding, the level of the INR and the indication for anticoagulation. For over-warfarinisation without bleeding, stoppage of warfarin with possible oral vitamin K administration is usually sufficient. However, if this is accompanied by significant bleeding, reversal of anticoagulation with factor replacement becomes necessary (Table 1).[7]
As for patients scheduled for surgery, anticoagulant reversal must be done before surgery to restore normal coagulation status.
Heparin
Heparin is an anticoagulant that works by binding to and potentiating the activity of antithrombin, which then inhibits thrombin. Heparin is used for the treatment and prevention of thrombosis.
High doses of heparin can cause severe bleeding. In this event, discontinuation of heparin is usually sufficient owing to its short half-life of 8 hours. If rapid reversal of heparin effect is required, protamine sulphate is very effective for unfractionated heparin, but only reverses ~60% of the antifactor Xa activity of low-molecular-weight heparin, and has negligible effects on fondaparinux and danaparoid (a mixture of anticoagulant glycosaminoglycans used to treat heparin-induced thrombocytopenia).[8,9]
Non-vitamin K antagonist oral anticoagulants
Non-vitamin K antagonist oral anticoagulants (NOACs) include thrombin inhibitors, e.g. dabigatran, and FXa inhibitors, e.g. rivaroxaban and apixaban. Outcomes of major bleeding are no worse than with vitamin K antagonists. Three NOAC reversal agents are in various stages of development, i.e. idarucizumab for thrombin inhibitors, andexanet for FXa inhibitors, and ciraparantag for all NOACs.[10]
Antiplatelet agents
Aspirin exerts its antiplatelet effect by irreversibly binding to the enzyme cyclo-oxygenase. Other antiplatelet agents include NSAIDs and adenosine diphosphate (ADP) receptor inhibitors, such as clopidogrel (Plavix). Mild bleeding and bruising may occur in response to trauma or surgery, but are likely to be exacerbated with coexisting medical conditions, such as haemophilia, renal disease and leukaemia. More severe spontaneous bleeds, e.g. from the gastrointestinal tract, occur less frequently. The effect of aspirin and clopidogrel lasts for 5 - 7 days, i.e. the entire lifespan of the platelet.
Abnormalities of platelet number
Platelet defects are typically associated with mucocutaneous bleeding, with the severity depending on the degree of the thrombocytopenia. In general, the risk of bleeding is low when platelets are >80 χ 109/L, and significantly increased when the platelet count is <20 χ 109/L (where spontaneous bleeding may occur). Platelet transfusion is indicated in all bleeding patients to maintain a platelet count of 50 - 100 χ 109/L (depending on the site of blood loss), as well as in selected patients with a platelet count <20 χ 109/L as bleeding prophylaxis. Causes of thrombocytopenia are divided into: (i) central (production failure); and (ii) peripheral (reduced survival). These are summarised in Table 2, and some of the more important causes are discussed below.
Immune thrombocytopenia
Immune thrombocytopenia (ITP) is an acquired, auto-immune disorder with the formation of antiplatelet antibodies against platelets and megakaryocytes, resulting in increased destruction and inadequate production of platelets.[11,12]
The term ITP refers to immune thrombocytopenia and no longer to the older term idiopathic thrombocytopenic purpura. The threshold for ITP and for clinical thrombocytopenia is defined as a platelet count <100 χ 109/L.[13]
The incidence of ITP is ~3 - 4.5/100 000/year.[14,15] In South Africa (SA), primary ITP predominantly affects young females.[16]
In Europe, however, the median age is 57 years, with a rising incidence with advancing age (>60 years) and a less marked gender difference.[14,15]
The presentation of ITP may be acute or insidious. Three phases of ITP are recognised: (i) newly diagnosed (0 - 3 months from diagnosis); (ii) persistent (3 - 12 months); and (iii) chronic (>12 months).[13 Most adult patients go on to develop chronic ITP. Spontaneous remissions occur in 5 - 11% of adults, mostly in the first year after diagnosis.[17 ITP may be primary (~80% of cases), with no identifiable cause, or secondary (~20% of cases), due to a number of causes. In SA, a paradigm shift has been noted, with an increasing number of patients with secondary ITP, largely due to HIV infection.[16]
Primary ITP is a diagnosis of exclusion. Secondary causes need to be excluded, including infections (e.g. HIV), auto-immune disorders (e.g. systemic lupus erythematosus), drugs (e.g. rifampicin, quinine and heparin), and lymphoproliferative disorders (e.g. chronic lymphocytic leukaemia, and lymphoma). In the classic patient with primary ITP (young female, isolated thrombocytopenia, no abnormalities on the peripheral smear such as fragments or atypical cells), a bone marrow aspirate and trephine biopsy (BMAT) is not indicated.[18] However, in patients with a suspected secondary cause, or in whom the presentation is atypical, or in individuals >60 years of age, a BMAT must be performed.
Patients with ITP may be asymptomatic (where the platelet count is usually >30 χ 109/L) or may present with bleeding, which is typically of the mucocutaneous type. The incidence of major bleeding events, such as intracranial haemorrhage and cavity bleeding, is low. The platelet count remains the best known predictor of bleeding events in ITP. Lymphadenopathy and hepatosplenomegaly are generally not encountered in primary ITP and, if present, indicate another cause or secondary ITP.
The decision to initiate treatment is primarily based on whether the patient is symptomatic (bleeding) and the level of the platelet count (<30 χ 109/L). The goal of treatment is to stop the bleeding and increase the platelet count to a safe level and not necessarily to achieve a normal platelet count.
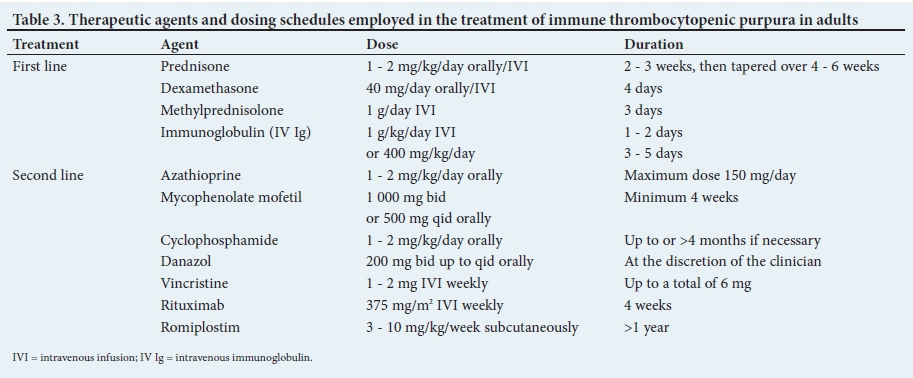
Therapeutic agents used in the treatment of ITP are presented in Table 3.
Corticosteroids (CS) are the mainstay of treatment in newly diagnosed ITP. Prednisone is the preferred initial treatment. Alternative CS include dexamethasone and methylprednisolone.
Platelet transfusions are reserved for patients with severe thrombocytopenia with active bleeding or recent onset of 'red purpura', such as oral haemorrhagic bullae. Platelet transfusion is not indicated in patients without bleeding, irrespective of the severity of the thrombocytopenia.
For emergency treatment, intravenous immunoglobulin (IV Ig) and IV/oral CS should be used in combination with platelet transfusions. Emergency splenectomy may rarely be necessary in such patients.
For persistent ITP, treatment options include CS and second-line immunosuppressive agents (such as azathioprine and mycophenolate mofetil) as steroid-sparing drugs. If these prove unsuccessful, other second-line agents, such as cyclophosphamide, danazol and vincristine, may be considered. Alternative drugs include rituximab and thrombopoietin-receptor agonists (TPO-RAs). The choice of second-line therapies depends on the experience of the clinician, efficacy and safety of the drug, availability, cost and patient preferences.
Chronic ITP refers to disease that continues for >12 months. It is likely that the patient has been on intermittent CS and/or other immunosuppressive or second-line therapy, with a variable clinical response. The three major treatment options for chronic ITP include splenectomy, TPO-RAs and rituximab. Each of these options has advantages and disadvantages and treatment must be individualised to the patient.
Splenectomy is the most definitive therapy for ITP and is effective for persistent and chronic ITP after failure of CS therapy. It is recommended that splenectomy be delayed for at least 6 months (preferably 12 months) from diagnosis, as there is a chance of spontaneous remission in 5 - 11% of cases.[17] The overall response rate is 70 - 90% (complete response 50 - 60%, partial response 20 -30%). The efficacy of open splenectomy and laparoscopic splenectomy is similar. However, laparoscopic splenectomy has fewer surgical complications, including less postoperative pain, earlier diet tolerance and shorter hospital stay. The risk of overwhelming post-splenectomy infection is increased 1.4-fold in the first year after the procedure. Vaccination against Streptococcus pneumoniae, Neisseria meningitides and Haemophilus influenzae is recommended 2 weeks prior to the procedure.[19,20] If relapse occurs post splenectomy, accessory splenic tissue (splenunculi) should be excluded. In two recent local studies it was indicated that splenectomy is beneficial and may be considered as the preferred second-line therapy in chronic ITP and failed CS therapy, especially in the SA public health sector.[16,21]
Two TPO-RAs are currently available for use in ITP, romiplostim and eltrombopag. These agents are effective in both splenectomised and non-splenectomised patients, with a response rate of up to 88%. To maintain durable remission, treatment is usually required for months to years before discontinuation. Currently, the high cost of TPO-RAs prohibits their use in the public health sector.[22,23]
Rituximab is an anti-CD20 monoclonal antibody, with an offlabel indication in patients with ITP.[24] Remission with rituximab occurs in up to two-thirds of patients but is durable in only one-third. However, the combination of rituximab and high-dose dexamethasone has shown a response rate of 71%, with a durable remission rate of 57%.[25] A higher risk of infection is anticipated with the use of rituximab.
Locally, there is an increase in secondary ITP, particularly in association with HIV.[16,21] The presentation of secondary ITP is identical to that of primary ITP, except for an increased likelihood of cytopenias and association of lymphadenopathy and hepatosplenomegaly.
The acute management of secondary ITP is identical to that of primary ITP, with the important proviso that the underlying cause be specifically treated (e.g. institution of antiretroviral therapy if patients are HIV-positive, removal of offending drug). With HIV, there is a potentially higher risk of infection, which may be exacerbated by immunosuppressive drugs and splenectomy. Although the duration to platelet recovery is slower in HIV-seropositive patients with ITP, the overall response to treatment is similar to that in the HIVnegative counterpart.[16] Splenectomy has been shown to be effective and safe, irrespective of the HIV status of the patient, and remains an appropriate second-line treatment for ITP.[21]
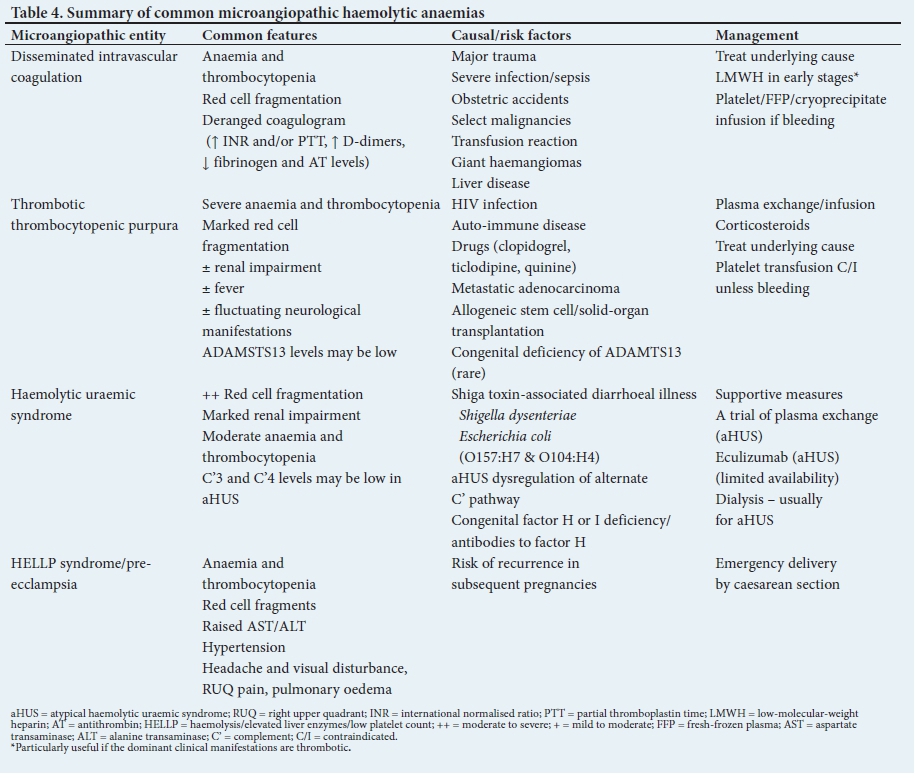
Micro-angiopathic haemolytic anaemia
Micro-angiopathic haemolytic anaemia (MAHA) encompasses a group of entities that are associated with red cell fragmentation haemolysis and thrombocytopenia (Table 4). Although MAHA may be complicated by thrombocytopenia-related blood loss, the risk of bleeding is significantly higher among patients with DIC. In contrast, bleeding is an unusual complication in thrombotic thrombocytopenic purpura (TTP), despite the thrombocytopenia often being very severe (<10 χ 109/L).
DIC is characterised by systemic activation of the coagulation cascade with production of microthrombi in the small vessels of multiple organs, resulting in organ dysfunction and consumption of coagulation factors and platelets.[26] It can manifest with bleeding and/or thromboembolism, depending on the rate of fibrinolysis and coagulation factor consumption relative to the compensatory production of these proteins. Causes of DIC include severe sepsis, obstetric calamities, major trauma and some malignancies (particularly acute promyelocytic leukaemia). All of these can result in systemic coagulation activation, either by exposing procoagulant proteins, or by generating procoagulant cytokines. It is diagnosed by demonstrating evidence of the following:
• consumption of blood clotting factors, leading to prolongation of the INR and/or PTT, with a decreasing fibrinogen level. Fibrinogen is, however, an acute-phase reactant, and is therefore not invariably low in patients with DIC
• consumption of anticoagulant molecules (such as antithrombin)
• accumulation of the products of fibrinolysis (such as D-dimers)
• a decreasing platelet count.
The abovementioned factors are diagnostic in an appropriate clinical setting.
Treatment of DIC is aimed at active management of the underlying cause, and symptomatic management of the associated organ and coagulation abnormalities. Treatment options include FFP and platelet transfusions in the event of bleeding, or low-molecular-weight heparin therapy when thromboembolic phenomena predominate. The use of antifibrinolytic agents is generally not advised owing to the risk of exacerbation of the microvascular thrombotic process, but may be of benefit in patients with hyperfibrinolysis (typified by very low fibrinogen and markedly elevated D-dimer levels). The mortality rate associated with DIC is high, particularly in the presence of pronounced organ dysfunction or severe coagulopathy.
Organ dysfunction
Liver disease
Coagulopathy in patients with liver disease can be difficult to differentiate from laboratory-determined DIC values. Haemostatic abnormalities due to liver disease include:
• thrombocytopenia due to thrombopoietin deficiency, hyper-splenism (if enlarged spleen) and possible megakaryocytic suppression (secondary to alcohol or hepatitis B or C infection)
• coagulopathy due to impaired production of clotting factors by the liver, vitamin K deficiency (e.g. alcohol abuse, obstructive jaundice), production of functionally abnormal fibrinogen (dysfibrinogenaemia)
• increased fibrin degradation products due to: (i) impaired hepatic clearance; and/or (ii) hyperfibrinolysis (impaired clearance of tissue plasminogen activator and decreased production of fibrinolytic inhibitors).
As there is a concomitant depletion of both pro- and anticoagulant molecules in patients with synthetic dysfunction of the liver, bleeding manifestations are often not as severe as would be anticipated from the degree of the laboratory derangements. However, associated renal dysfunction or infection can predispose to bleeding. In particular, gastrointestinal bleeding from oesophageal varices is a concern in patients with portal hypertension, requiring reduction of the portal pressure and ligation of the varices. Coagulation factor, fibrinogen and platelet replacement therapy may be needed, but caution should be exercised against liberal use of FFP in liver disease, as the plasma volume expansion may elevate portal pressure and thereby paradoxically increase the risk of variceal bleeding.[27] Bleeding due to hyperfibrinolysis, diagnosed with viscoelastic tests such as the thromboelastogram (TEG), can respond to antifibrinolytic agents, e.g. tranexamic acid.
Renal disease
Numerous haemostatic disturbances are observed in renal disease, which may predispose to a hypo- or hypercoagulable state. There is no superior pathogenic factor to determine whether a patient would be prone to bleeding or thrombosis, where the dynamics of events are often influenced by comorbid factors.[28] Tendency to bleed is caused by platelet dysfunction (due to accumulation of toxic metabolites, fibrinogen degradation products, anaemia, drugs, etc.) and decreased FXI/XII level. Desmopressin and/or antifibrinolytics are generally effective in controlling uraemic bleeding.
Vascular defects
Acquired vascular bleeding disorders include the following:
Scurvy. Vitamin C promotes peptidyl hydroxylation of procollagen, and its deficiency causes abnormal collagen formation with defective perivascular support. This predisposes to capillary fragility and mucocutaneous bleeding. Treatment is with vitamin C 200 mg daily.
Henoch-Schönlein purpura. This idiopathic disorder is primarily a disease of children, but may occur at any age, and is characterised by abdominal colic, arthritis, nephritis and palpable purpura. Biopsy of the skin shows an acute immune-related vasculitis and complement/ immunoglobulin complexes. Treatment entails supportive care and steroids in more severe cases.
Paraproteinaemia and amyloidosis. The mechanism of bleeding is multifactorial, including interference with coagulation factor levels/ function, impaired platelet aggregation and deposits of light chain/ amyloid fibrils in cutaneous blood vessels, with increased vessel fragility. Cryoglobulins may similarly deposit in dermal vessels and cause vasculitis and purpura. Management entails treatment of the underlying condition.
Senile purpura. In the elderly there is loss of subcutaneous collagen and elastin fibres. Bruising is usually induced by minor trauma.
Conclusion
Acquired bleeding disorders encompass a heterogeneous group of conditions with varied aetiologies. A detailed history and complete physical examination are imperative for meaningful interpretation of laboratory tests and appropriate treatment. Bleeding is often a presenting manifestation of systemic disease and therefore necessitates a multidisciplinary team approach.
Acknowledgements. None.
Author contributions. NA: composed subsection and co-ordinated manuscript compilation; JV: composed subsection and manuscript review; SL: composed subsection and manuscript review; MP: composed subsection and manuscript review; and SM: composed subsection.
Funding. None.
Conflicts of interest. None.
References
1. Alli N, Vaughan J, Louw S, Schapkaitz E, Mahlangu J. Inherited bleeding disorders. S Afr Med J 2018;108(1):9-15. https://doi.org/10.7196/SAMJ.2018.v108i1.13020, [ Links ]
2. Kershaw G, Favaloro EJ. Laboratory identification of factor inhibitors: An update. Pathology 2012;44(4):293-302. https://doi.org/10.1097/PAT.0b013e328353254d [ Links ]
3. Lai JD, Lillicrap D. Factor VIII inhibitors: Advances in basic and translational science. Int J Lab Hematol 2017;39(Suppl 1):6-13. https://doi.org/10.1111/ijlh.12659 [ Links ]
4. Wang M, Cyhaniuk A, Cooper DL, Iyer NN. Identification of people with acquired hemophilia in a large electronic health record database. J Blood Med 2017;8:89-97. https://doi.org/10.2147/JBM.S1360605 [ Links ]
5. Kruse-Jarres R, Kempton CL, Baudo F, et al Acquired hemophilia A: Updated review of evidence and treatment guidance. Am J Hematol 2017;92(7):695-705. https://doi.org/10.1002/ajh.24777 [ Links ]
6. Oldenburg J, Zeitler H, Pavlova A. Genetic markers in acquired haemophilia. Haemophilia 2010;16(Suppl 3):41-45. https://doi.org/10.1111/j.1365-2516.2010.02259 [ Links ]
7. Jacobson BF, Schapkaitz E, Haas S, et al. Maintenance of warfarin therapy at an anticoagulation clinic. S Afr Med J 2007;97(12):1259-1265. https://doi.org/10.7196/SAMJ.194 [ Links ]
8. Jacobson BF, Louw S, Buller H, et al. Venous thromboembolism: Prophylactic and therapeutic practice guideline. S Afr Med J 2013;103(4):261-267. https://doi.org/10.7196/samj.6706 [ Links ]
9. Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002;49(6):S11-S25. [ Links ]
10. Levy JH, Douketis JD, Weitz JI. Reversal agents for non-vitamin K antagonist oral anticoagulants. Nat Rev Cardiol 2018 (epub ahead of print). https://doi.org/10.1038/nrcardio.2017.223 [ Links ]
11. Cines DB, McMillan R. Pathogenesis of chronic immune thrombocytopenic purpura. Curr Opin Hematol 2007;14(5):511-514. https://doi.org/10.1097/MOH.0b013e3282ba5552 [ Links ]
12. Olsson B, Anderson PO, Jernas M, et al. T-cell mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nature Med 2003;9(9):1123-1124. https://doi.org/10.1038/nm921 [ Links ]
13. Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood 2009;113(11):2386-2393. https://doi.org/10.1182/blood-2008-07-162503 [ Links ]
14. Frederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood 1999;94(3):900-913. [ Links ]
15. Neylon AJ, Saunders PW, Howard MR, et al. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: A prospective study of a population-based cohort of 245 patients. Br J Haematol 2003;122(6):966-974. https://doi.org/10.1046/j.1365-2141.2003.04547 [ Links ]
16. Variava F. Immune thrombocytopenia at Chris Hani Baragwanath Academic Hospital. MMed dissertation. Johannesburg: University of the Witwatersrand, 2014. http://wiredspace.wits.ac.za/jspui/bitstream/10539/18647/1/ITP%20at%20CHB.pdf (accessed 6 February 2018). [ Links ]
17. Stasi R, Stipa E, Masi M, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 1995;98(5):436-442. [ Links ]
18. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117(16):4190-4207. https://doi.org/10.1182/blood-2010-08-302984 [ Links ]
19. Ghanima W, Godeau B, Cines D, et al. How I treat immune thrombocytopenia: The choice between splenectomy or a medical therapy as a second-line treatment. Blood 2012;120(5):960-969. https://doi.org/10.1182/blood-2011-12-309153 [ Links ]
20. Cordera F, Hall Long K, Nagorney DM, et al. Open versus laparoscopic splenectomy for idiopathic thrombocytopenic purpura: Clinical and economic analysis. Surgery 2003;134:45-52. https://doi.org/10.1067/msy.2003.204 [ Links ]
21. Antel KR, Panieri E, Novitzky N. Role of splenectomy for immune thrombocytopenic purpura (ITP) in the era of new second-line therapies and in the setting of a high prevalence of HIV-associated ITP. S Afr Med J 2015;105(4):408-412. https://doi.org/10.7196/samj.8987 [ Links ]
22. Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: A double-blind randomised controlled trial. Lancet 2008;371(9610):395-403. https://doi.org/10.1016/S0140-6736(08)60203-2 [ Links ]
23. Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltombopag: Results from the EXTEND study. Blood 2009;114(22):682 [ Links ]
24. Auger S, Duny Y, Rossi JF, et al Rituximab before splenectomy in adults with primary immune thrombocytopenic purpura: A meta-analysis. Br J Haematol 2012;158(3):386-398. https://doi.org/10.1111/j.1365-2141.2012.09169 [ Links ]
25. Ghanima W, Elstrom R, Bussel JB. The combination of three dexamethasone cycles and rituximab yields high response rate in previously treated immune thrombocytopenia (ITP). Haematologica 2011;96:95. [ Links ]
26. Wada H, Matsumoto T, Yamashita Y. Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. J Intens Care 2014;2(1):15. https://doi.org/doi.org/10.1186/2052-0492-2-15.2 [ Links ]
27. Kujovich JL. Coagulopathy in liver disease: A balancing act. ASH Hematol Educ Program 2015;2015(1):243-249. https://doi.org/doi.org/10.1182/asheducation-2015.1.243 [ Links ]
28. Pavord S, Myers B. Bleeding and thrombotic complications of kidney disease. Blood Rev 2011;25:271-278. https://doi.org//10.1097/01.ASN/0000081661.10246.33 [ Links ]
 Correspondence:
Correspondence:
N Alli
nazeer.alli@nhls.ac.za
Accepted 2 February 2018


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}