










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.108 no.2 Pretoria Fev. 2018
http://dx.doi.org/10.7196/samj.2018.v108i2.12579
RESEARCH
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency in South Africa
Y GanieI, II; C AldousIII; Y BalakrishnaIV; R WiersmaV
IFCP (Paed), Cert Endocrinology and Metabolism (SA) Paed; Department of Paediatrics and Child Health, Nelson R Mandela School of Medicine, College of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
IIFCP (Paed), Cert Endocrinology and Metabolism (SA) Paed; Division of Paediatric Endocrinology, Inkosi Albert Luthuli Central Hospital, KwaZulu-Natal, Durban, South Africa
IIIPhD; Department of Internal Medicine, Nelson R Mandela School of Medicine, College of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
IVMSc; Biostatistics Unit, South African Medical Research Council, Durban, South Africa
VPhD; Department of Paediatric Surgery, Inkosi Albert Luthuli Central Hospital and Nelson R Mandela School of Medicine, College of Health Sciences, University of KwaZulu-Natal Durban, South Africa
ABSTRACT
BACKGROUND: Congenital adrenal hyperplasia (CAH) caused by deficiency of the 21-hydoxylase (21-OH) enzyme is the most common form
of CAH worldwide.
OBJECTIVE: To evaluate the prevalence of CAH due to 21-OH deficiency, and its clinical presentation and biochemical profiles in affected children.
METHODS: We performed a retrospective subset analysis of 44 children with confirmed CAH.
RESULTS: All the children had classic CAH. The majority (59.8%) had classic salt-wasting (CSW) CAH and 40.1% had simple virilising (SV) CAH. The median age of presentation was 8.1 years (interquartile range (IQR) 4.5 - 11) in the SV group and 2 months (IQR 2 weeks - 5 months) in the CSW group (p=0.0001). No difference in age of presentation was noted between males and females (p=0.541). The clinical presentation was significantly different between the CSW and SV groups, and between males and females in the CSW group (p<0.0001). Most of the females with 46,XX CSW CAH (66.7%) presented with disorders of sex development (DSD), while the remaining 33.3% presented with DSD and dehydration and shock. All the males with 46,XY CSW CAH presented with dehydration and shock. Overall, 37.9% (11/29) of the children were obese or overweight at presentation. Gonadotrophin-releasing hormone-dependent central precocious puberty was observed on follow-up in 29.4% (10/34) of the children at a median of 6.7 years (IQR 5 - 7.7).
CONCLUSION: The diagnosis of CAH is delayed in males and females in both SV and CSW forms of the disorder, which probably contributes to under-reporting of cases and a high mortality rate.
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterised by enzyme deficiencies that impair adrenal steroid biosynthesis. The most common form is deficiency of the 21-hydroxylase (21-OH) enzyme, accounting for >90% of cases. The incidence ranges from 1:10 000 to 1:20 000 live births.[1]
There are three types of CAH, depending on the severity of the enzyme deficiency. The most common type is the classic salt-wasting (CSW) form, occurring in 75% of cases, which is characterised by cortisol and aldosterone deficiency and androgen excess. The simple virilising (SV) type is characterised by cortisol deficiency and androgen excess but adequate amounts of aldosterone. In both CSW and SV CAH, the excess androgens cause virilisation of the genitalia in girls, leading to disorders of sex development (DSD). The cortisol deficiency may present with an adrenal crisis, while the aldosterone deficiency causes sodium and water loss resulting in dehydration, shock and electrolyte abnormalities. In addition to the CSW and SV forms of CAH, there is also a non-classic form (NCCAH), which is characterised by mild deficiency of the 21-OH enzyme with variable degrees of postnatal androgen excess but is sometimes asymptomatic. Girls with the NCCAH form are therefore not virilised at birth and do not present with DSD. There is mild subclinical impairment of cortisol in NCCAH that does not lead to adrenal crises. NC forms of CAH are more common than classic forms, occurring in 1 - 3% of the general population, and are often misdiagnosed as precocious pubarche or polycystic ovarian disease in young women.[1] Newborn screening in developed countries has reduced the morbidity and mortality associated with CAH.[2]
There is a lack of reporting on the prevalence of CAH in resource-poor countries such as South Africa (SA), affecting diagnosis, treatment and management outcomes. Lack of screening facilities results in delayed and missed diagnosis and a high mortality in salt-wasting forms.
In 1976, Geefhuysen et al.[3]reported that CAH was less common in black compared with white SA children. We recently reported that CAH was the third most common diagnosis in children with DSD attending a specialised endocrine unit.[4] In 2008, Osifo and Nwashilliv[5] retrospectively reported on 27 children with CAH in Nigeria, highlighting the challenges of managing these children in a developing country. Tayel et al.[6] reported a high prevalence of CAH in Alexandria, Egypt, justifying the need for a newborn screening programme.
In a large retrospective population-based cohort study in Sweden,[7] the authors concluded that diagnoses in boys and girls with CAH were equally missed and that neonatal screening improved detection rates. However, in other European studies in which CAH was diagnosed clinically, the diagnosis was missed in males more than in females.[8-10]
Objective
Retrospective evaluation of the prevalence, clinical presentation and biochemical profiles of CAH caused by 21-OH deficiency in children attending a tertiary hospital in Durban, SA.
Methods
A database of patients with DSD presenting to the endocrine unit at Inkosi Albert Luthuli Central Hospital (IALCH) in Durban has been maintained from January 1995. This database was used to analyse the past records of patients with DSD. The database obtained class approval from the Biomedical Research and Ethics Council (ref. no. BE479/14) in 2015.
IALCH is a provincial tertiary hospital in KwaZulu-Natal Province (KZN) and serves a population of ~5 million children from 11 health districts. It is the only referral centre for paediatric patients with endocrine problems in KZN. Children are initially evaluated at their district or regional hospitals and referred to IALCH for subspecialist support. This is a retrospective review of a subset of children with a diagnosis of CAH made by the paediatric endocrinologist, based on clinical presentation and confirmed with biochemical investigations.
The following information was recorded in the database: age at presentation, birth weight, demographics including residential areas and referral hospitals, sex of rearing, gender dysphoria, consanguinity and a family history of DSD. Clinical symptoms and presentation of the patients were also recorded. Patients were noted as presenting with DSD alone, DSD with dehydration and shock, dehydration and shock without DSD, precocious puberty or amenorrhoea.
Clinical findings at presentation, including anthropometry (height or length, weight and body mass index (BMI) z-scores), were recorded. The patients' nutritional status was classified using the World Health Organization (WHO) child growth reference standards.[11] Overweight was defined as a z-score >1 SDS (standard deviation score) and obesity as a z-score >2 SDS above the WHO growth reference median. Pubertal stage was assessed using the Tanner scale. The Prader scoring system was used to determine the degree of external virilisation.[12]
Karyotyping was performed in all patients on peripheral blood leucocytes. The biochemical diagnosis of 21-OH deficiency was confirmed by hormonal evaluation. The patients' biochemical values (serum electrolytes and blood glucose) and levels of sex steroids at presentation, before the initiation of hormone replacement, were measured. Gonadotrophins, 17-hydroxyprogesterone (17-OHP), dehydroepiandrostenedione (DHEA-S), androstenedione, testosterone and oestradiol were recorded. All hormone levels were assayed with commercially available kits. Sex steroids and adrenocorticotrophic hormone (ACTH) were measured using chemiluminescence radioimmunoassay (Immulite 2000, Siemens, USA) in a central laboratory at IALCH. Plasma renin (mass) was measured by Cisbio radioimmunoassay (Cisbio, France), and serum aldosterone and 17-OHP were measured by Beckman coulter radioimmunoassay (Beckman Coulter, USA). Results were quantified using the Cobra II gamma counter (Packard, USA). Reference values are highly method dependent. Our unit has established its own normal values in conjunction with the local and regional laboratories. Values were compared with age- and sex-matched data from unaffected children.
An early-morning (before 08h00) baseline serum 17-OHP level is the screening test of choice in symptomatic individuals after infancy.
Reference ranges of 17-OHP were 0.2 - 2.3 nmol/L for newborns and infants, 0.1 - 2.7 nmol/L for prepubertal children, 0.1 - 5.4 nmol/L for pubertal boys, 0.1 - 8 nmol/L for pubertal girls, 0.4 - 2.1 nmol/L for post-menarche girls during the follicular phase, and 1.0 - 8.7 nmol/L for post-menarche girls during the luteal phase.[13,14]
A screening serum 17-OHP level of >6 nmol/L was an indication to obtain a complete adrenocortical profile and to perform a high-dose (250 ug) cosyntropin stimulation test (HDSST). The HDSST is the gold standard to differentiate 21-OH deficiency from other enzyme defects and to confirm the diagnosis in borderline cases.[15]
Levels of 17-OHP and cortisol were measured at baseline, then 30 and 60 minutes after the administration of cosyntropin. The plasma cortisol level at 30 minutes should be >550 nmol/L. A normal response to cosyntropin is an incremental increase of <10 nmol/L above basal concentration at 60 minutes. Patients with NC CAH show an incremental increase of >20 nmol/L, while heterozygote CAH patients have an intermediate response with considerable overlap into the normal range.[15] The 17-OHP values were plotted on a nomogram to determine the severity of the disease.[16] Unfortunately cosyntropin was not easily available, and we were unable to perform the test on an urgent basis in some patients. These patients were treated according to the clinical findings and supportive baseline biochemical results. Molecular genetic analysis is also not routinely available for patients with DSD and CAH in SA.
Findings on radiological investigations, including the findings of a pelvic ultrasound scan, and bone age were also captured.
Patients were divided into CSW and SV groups, and further subdivided according to karyotypes. The clinical characteristics and biochemical values were compared between and within the groups.
The diagnosis of central precocious puberty (CPP) was made clinically and was supported by biochemical findings. Puberty that begins before 8 years of age in girls and before 9 years of age in boys is defined as precocious puberty. In girls, the first sign of puberty is usually breast enlargement, which may initially be unilateral. Pubic and axillary hair may appear before, at about the same time as, or well after the appearance of breast tissue. Menarche is a late event and does not usually occur until 2 - 3 years after onset of breast enlargement. In boys, the earliest evidence of puberty is testicular enlargement (>4 mL). This is followed by growth of the penis and scrotum, which typically occurs at least a year after testicular enlargement. Measurement of serum testosterone is useful in boys with suspected precocious puberty. A luteinising hormone level of <0.1 IU/L is generally prepubertal. Random follicle-stimulating hormone levels do not discriminate between prepubertal and pubertal children.
Statistical analysis
Data were analysed using Stata version 14 (StataCorp, USA). Categorical variables were described using frequencies and percentages, and associations between groups were tested using Pearson's x2 test or Fisher's exact test, where applicable. Continuous variables were described using medians with interquartile ranges (IQRs), and differences between groups were tested using the Kruskal-Wallis test. Spearman correlations were computed between age and sex steroids. Results were considered statistically significant for p-values <0.05.
Results
A total of 3 233 patients attended the endocrine clinic over the review period, and 44 (1.4%; 95% confidence interval 0.99 - 1.82) had a diagnosis of CAH. Thirty-nine patients were diagnosed between the years 2006 and 2016.
All the patients (N=44) were diagnosed with classic CAH (Table 1). The majority (59.8%, n=26) had CSW CAH, and 40.9% (n=18) had SV CAH. Overall, there were more females (80.8%) than males (19.2%) among the classic CAH patients. There was no significant gender difference between the CSW and SV groups (p=0.999).
Age at presentation
The diagnosis was established significantly later in the SV CAH group (median 8.1 years (IQR 4.5 - 11)) than in the CSW CAH group (median 2 months (IQR 2 weeks - 5 months)) (p=0.0001). The median age at presentation in the 46,XY CSW group was 3 months (IQR 3 - 4), while that in the 46,XX CSW group was 1 month (IQR 2 -5) (p=0.241). In the 46,XY SV group the median age of presentation was 7 years (IQR 5.5 - 9.5), and in the 46,XX SV group it was 9.2 years (IQR 4.3 - 10.8) (p=0.832). There was no significant difference in median age of presentation between females and males (p=0.541) in the CSW and SV groups (Fig. 1).

Baseline demographic characteristics
Baseline and demographic characteristics of the cohort are summarised in Table 1. There was no family history of consanguinity among the patients with CAH. A positive family history of CAH was noted in 13.6% of patients (n=6), or three families. In the first family, the sibling pair each had 46,XX CSW CAH, while in the second family, the siblings had 46,XX SV CAH. In the third family, two siblings were diagnosed with CAH; however, the older sibling had 46,XY SV CAH and the younger sibling 46,XX CSW CAH.
Baseline clinical characteristics
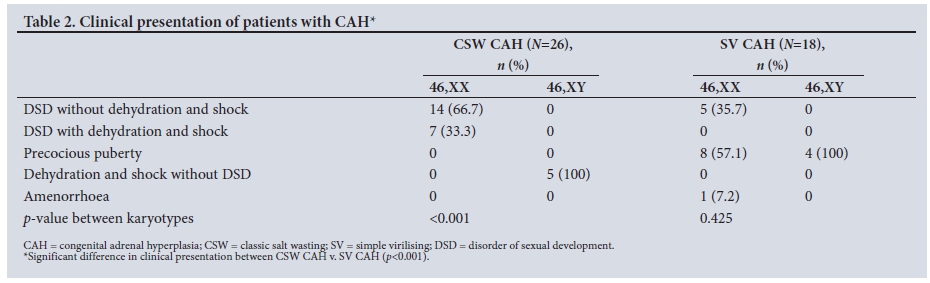
The clinical presentation was substantially different between the two groups (p<0.001) (Table 2). In the CSW group, 14/21 patients (66.6%) presented with DSD without dehydration and shock, while 7/21 (33.3%) presented with DSD and dehydration and shock. This difference was also significant between the males and females in the CSW group (p<0.001). Most of the females with 46,XX CSW (66.6%) were noted to have DSD on routine neonatal examination. None of the males with 46,XY CSW presented with DSD in the neonatal period. All the males (n=5) presented with dehydration and shock. In the SV group, 12 patients (66.7%) presented with precocious puberty and 5/14 females (35.7%) presented with DSD. The clinical presentation between male and females in the SV CAH group did not differ significantly (p=0.425).
Birth weight was recorded in 41 patients. More patients in the CSW group were of low birth weight compared with the SV group (p=0.039). No association was found between karyotype and birth weights within the two groups.
Anthropometric data were available for 29 patients (65.9%). Overall, 37.9% (n=11) of these were either obese or overweight. All the obese patients were black females. Compared with SV patients, notably more CSW patients (62.1%, n=18) had severe acute malnutrition. SV patients were more likely to be obese (n=5, 45.5%) or overweight (n=6, 54.5%) at presentation (p<0.001).
The median total daily dose of hydrocortisone was 15 mg/m2/d (IQR 10 - 16) in the CSW group and 12 mg/m2/d (IQR 11 - 15) in the SV group (p=0.251).
Baseline biochemical findings
There was a significant difference in serum sodium, potassium, bicarbonate, renin, aldosterone and DHEA-S levels between the CSW and SV groups (Table 3). The most significant difference between the two groups was in serum potassium and renin levels (p<0.0001). There was a significant negative correlation between sex steroids and age in the CSW group (Table 4). The younger patients in the CSW group tended to have higher sex steroid levels. No significant correlation was found in the SV group except for DHEA-S levels, which increased with age (p<0.008).

High-dose cosyntropin stimulation test
The HDSST test was performed in 20 (45.5%) of the patients (9 in the CSW group and 11 in the SV group). The median age at which the test was done was 3 months (IQR 1 - 6) in the CSW group and 7.5 years (IQR 4.8 - 9.7) in the SV group (p=0.002). There was no significant difference in serum 17-OHP and cortisol levels at baseline and at 30 and 60 minutes between the two groups. The maximum serum
17-OHP level (noted at 30 minutes) was 1 057 mmol/L (IQR 664 -
1 450) in the CSW group and 286.5 mmol/L (IQR 186 - 387) in the SV group. The serum cortisol level at 30 minutes was 222.5 mmol/L (140 - 368) in the CSW group and 160 mmol/L (145 - 221) in the SV group, confirming cortisol deficiency.
Central precocious puberty
Gonadotrophin-releasing hormone (GnRH)-dependent CPP was observed on follow-up in 10/34 patients (29.4%) at a median of 6.4 years (IQR 6.2 - 7.7) in girls and 7 years (IQR 6.7 - 7.4) years in boys (p=0.602). CPP was observed in five patients with SV CAH and five patients with CSW CAH. Of the patients with CPP, 60.0% had the female karyotype. There was no association between CPP and age at presentation, total daily dose of hydrocortisone, baseline gonadotrophins and sex steroids compared with patients who did not develop CPP.
Discussion
In this retrospective review over more than 20 years, we have shown that CAH caused by a deficiency of the 21-OH enzyme is not as common as might be expected among black children in KZN, SA.
All the patients had classic CAH and none had the mild NC form. Although NC forms of CAH are more prevalent than classic CAH in the general population, they present later in adolescence and adulthood. In our unit at IALCH, as per hospital policy, patients aged >12 years are referred to the adult units for management, which would explain the lack of patients with NCCAH.
As expected, CSW CAH presented significantly earlier than SV CAH. Overall there were fewer males than females, which could reflect unreported deaths in males in whom the diagnosis of CAH was missed. Although females with CSW CAH presented earlier than males, the median age of presentation was not significantly different between the two groups. This is similar to findings from the large population-based cohort study in Sweden,[7] but contrasts with reports from most studies in Europe in the pre-screening era, where females with CAH presented significantly earlier than males.[8-10] In resource-poor settings, especially in the absence of neonatal screening, examination of the genitalia is crucial in detecting children with CAH. Virilisation of the external genitalia without palpable gonads should arouse the suspicion of CAH and is an indication for prompt and emergency referral to an endocrine specialist.
It is known that female infants with CAH present with life-threatening adrenal crises in the second week, and males by the third week.[8] Although two-thirds of females in our study were diagnosed with DSD in the neonatal period, a third with DSD were missed or not recognised and subsequently presented with dehydration, salt loss and shock. All the males in the CSW group presented with dehydration and shock. Males have a lack of genital ambiguity that can lead to a delayed or missed diagnosis. However, females have virilisation of the external genitalia and should be recognised at birth. The death rate in CSW CAH without screening is 4 - 10%. Early diagnosis and treatment can prevent this mortality and morbidity, which has been averted in developed countries, where screening for CAH is universal.[14] Neonatal screening for 21-OH deficiency is not routinely available in SA.
There was a significant difference in growth parameters between the CSW and SV CAH groups. In the CSW CAH children, ongoing salt loss and dehydration results in malnutrition. Failure to thrive has previously been described in CSW CAH.[17] In contrast, all the patients in the SV group were either overweight or obese, and specifically all black females were obese. The majority of the patients in the SV group presented with precocious puberty. An increase in BMI has been associated with early puberty.[18] In some studies, the association is stronger in white girls than in black girls. However, obesity is not clearly associated with early puberty in boys or girls. The prevalence of childhood overweight and obesity in SA is one of the highest in Africa. The high frequency of obesity in the SV group may therefore be an indication of the background obesity epidemic in SA.
Both CSW and SV CAH are associated with impaired cortisol synthesis. The 21-OH enzyme converts 17-hydroxyprogesterone to 11-deoxycortisol and progesterone to deoxycorticosterone, which are precursors for cortisol and aldosterone synthesis. In CSW CAH characterised by a severe deficiency of the 21-OH enzyme, both cortisol and aldosterone are deficient. The biochemical profile is characterised by hyponatraemia, hyperkalaemia, low serum cortisol and aldosterone and high plasma renin and ACTH. Gastroenteritis is common in SA and can present with similar electrolyte abnormalities to CAH, which can mask diagnosis of the latter. Healthcare professionals should therefore have a high index of suspicion of CAH in children who present with hyponatraemia and hyperkalaemia. In SV CAH, however, serum sodium, potassium, aldosterone and renin levels are normal.[1] The finding of significantly different plasma renin and potassium levels between the groups is therefore to be expected and is secondary to the aldosterone deficiency in the CSW group.
Adrenal sex steroids, especially androstenedione and to a lesser extent testosterone, are elevated in 21-OH deficiency owing to the accumulation of cortisol precursors that are diverted to sex hormone synthesis. There was no significant difference in testosterone and androstenedione levels between the two groups in our study, but there was a significant difference in DHEA-S levels. This can be explained by age-related differences between the two groups, as serum levels of DHEA-S increase with age and the mean age was higher in the SV group. The back-door pathway is also more dominant in SW CAH, hence the lower DHEA-S levels.[19] DHEA-S is not a good marker of 21-OH activity, as it binds with high-affinity albumin, has a long plasma half-life and is not responsive to acute change or to ACTH stimulation.[1] The younger patients in the CSW group tended to have higher sex steroid levels than the older patients in the SV group, reflecting the mini-puberty of infancy.
While patients with SV CAH are known to present with peripheral precocious puberty, CPP caused by premature activation of the hypothalamic-pituitary gonadal axis is also described in patients with CAH. The CPP is triggered by the chronic mild to moderate intermittent elevation of androgens in patients who are non-compliant with medication. SV boys with advanced bone age of >12 years may have spontaneous CPP when treatment with hydrocortisone is initiated. The hydrocortisone suppresses the production of adrenal androgens and stimulates the release of gonadotrophins if hypothalamic maturation is already present.[20,21] All our patients with CPP are receiving treatment with a GnRH analogue. In SA, considerable limitations exist in optimally managing this condition. These include cultural and social issues, such as stigma and shame associated with genital abnormalities, that affect adherence to treatment.
Study limitations
This is a retrospective descriptive study, which is accompanied by the usual issues of data integrity such as missing data. To our knowledge, however, this review is the first and largest study evaluating the prevalence, clinical presentation and biochemical profiles in children with CAH due to 21-OH deficiency in SA. The information generated has important implications, as it can be used to develop guidelines including a neonatal screening programme for CAH to prevent deaths from adrenal crisis in unrecognised patients.
Conclusion
There are limited published data on CAH in SA. CAH caused by 21-OH deficiency appears to be infrequent in black SA children. In our cohort, the diagnosis of CAH was delayed in males and females in both the SV and CSW groups. One-third of females with genital ambiguity were missed at birth and presented with life-threatening adrenal crises. This missed and delayed diagnosis probably results in a high mortality rate and under-reporting of cases. In the absence of neonatal screening for CAH in SA, there is an urgent need to educate and train healthcare workers to recognise neonates with DSD early and refer them timeously for specialist care.
Acknowledgements. None.
Author contributions. All the authors accept responsibility for the entire content of the submitted manuscript and approved its submission.
Funding. None.
Conflicts of interest None.
References
1. Speiser WP. Congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Endocrinol Metab Clin North Am 2001;30(1):31-59. https://doi.org/10.1016/S0889-8529(08)70018-5 [ Links ]
2. Therell BL. Newborn screening for congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2001;30(1):15-30. https://doi.org/10.1016/S0889-8529(08)70017-3 [ Links ]
3. Geefhuysen J, Hall DMB, Worlfsdorf J. Congenital adrenal hyperplasia in blacks. S Afr Med J 1976;50(22):851-853. [ Links ]
4. Ganie Y, Aldous C, Balakrishna Y, Wiersma R. Disorders of sex development in children in KwaZulu- Natal Durban South Africa: 20-year experience in a tertiary centre. J Pediatr Endocrinol Metab 2017;30(1):11-18. https://doi.org/10.1515/jpem-2016-0152 [ Links ]
5. Osifo OD, Nwashilliv NJ. Congenital adrenal hyperplasia: The challenges of management in a developing country. Afr J Urol 2008;14(3):138-142. Retrieved from https://www.ajol.info/index.php/aju/article/view/8241 (accessed 2 January 2018). [ Links ]
6. Tayel SM, Ismael H, Kandil H, Abd Rabuh A, Sallam H. Congenital adrenal hyperplasia in Alexandria, Egypt: High prevalence justifying the need for a community based newborn screening program. J Tropic Pediatr 2011;57(3):232-234. https://doi.org/10.1093/tropej/fmq064 [ Links ]
7. Gildof S, Falhammar H, Guthenberg C, et al. One hundred years of congenital adrenal hyperplasia in Sweden: A retrospective population based cohort study. JAMA Pediatr 2014;168(6):567-574. https://doi.org/10.1001/jamapediatrics.2013.5321 [ Links ]
8. Phyllis WS, Ricardo A, Laurence SB, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2010;95(9):4133-4160. https://doi.org/10.1210/jc.2009-2631 [ Links ]
9. Lebovitz RM, Pauli RM, Laxova R. Delayed diagnosis in congenital adrenal hyperplasia: Need for newborn screening. Am J Dis Child 1984;138(6):571-573. https://doi.org/10.1001/archpedi.1984.02140440055014 [ Links ]
10. Kovács J, Votava F, Heinze G, Sólyom J, et al. Lessons from 30 years of clinical diagnosis and treatment of congenital adrenal hyperplasia in five middle European countries. J Clin Endocrinol Metab 2001;86(7):2958-2964. https://doi.org/10.1210/jcem.86.7.7638 [ Links ]
11. World Health Organization. Child Growth Standards. Geneva: WHO, 2017. http://www.who.int/childgrowth/en/ (accessed 16 January 2016). [ Links ]
12. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000;21(3):245-291. https://doi.org/10.1210/edrv.2L3.0398 [ Links ]
13. Hughes IA, Riad-Fahmy D, Griffiths K. Plasma 17OH-progesterone concentrations in newborn infants. Arch Dis Child 1979;54(5):347-349. [ Links ]
14. Orth D, Kovacs D, DeBold C. The adrenal cortex. In: Wilson JH, Foster DW, eds. Williams Textbook of Endocrinology. 8th ed. Philadelphia: WB Saunders, 1992. [ Links ]
15. Lashansky G, Saenger P, Fishman K, et al. Normative data for adrenal steroidgenesis in a healthy paediatric population: Age and sex related changes after ACTH stimulation. J Clin Endocrinol Metab 1991;73(3):674-686. https://doi.org/10.1210/jcem-73-3-674 [ Links ]
16. New MI, Lorenzen F, Lerner AJ, et al. Genotyping steroid 21-hydroxylase deficiency: Hormonal reference data. J Clin Endocrinol Metab 1983;57(2):320-326. https://doi.org/10.1210/jcem-57-2-320 [ Links ]
17. Sopfe J, Simmons JH. Failure to thrive, hyponatremia, and hyperkalemia in a neonate. Pediatr Ann 2013;42(5):74-79. https://doi.org/10.3928/00904481-20130426-09 [ Links ]
18. Mamun AA, Hayatbakhsh MR, O'Callaghan M, Williams G, Nauman J. Early overweight and pubertal maturation - pathways of association with young adults' overweight: A longitudinal study. Int J Obes (Lond) 2009;33(1):14-20. https://doi.org/10.1038/ijo.2008.220 [ Links ]
19. Turcu AF, Auchus RJ. Adrenal steroidgenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2015;44(2):275-296. https://doi.org/10.1016/j.ecl.2015.02.002 [ Links ]
20. Oyenusi EE, Oduwole AO, Kooris OO, Ademola AO, Nwaogu NT, Sabotages SB. Congenital adrenal hyperplasia at the Lagos University Teaching Hospital: A 10-year review. J Clin Sci 2016;13(4):178-186. https://doi.org/10.4103/2468-6859.192291 [ Links ]
21. Guven A, Cebeci N, Hancili S. Gonadotropin releasing hormone analogue treatment in children with congenital adrenal hyperplasia complicated by central precocious puberty. Hormones (Athens) 2015;14(2):265-271. https://doi.org/10.14310/horm.2002.1555 [ Links ]
22. Lesley J, Tetlow, Clayton P, Brook C. Tests and normal values in pediatric endocrinology. In: Brook C, Clayton P, Brown R, eds. Brook's Clinical Pediatric Endocrinology. 5th ed. Oxford. Blackwell Publishing, 2005:523-564. [ Links ]
 Correspondence:
Correspondence:
Y Ganie
yasmeen.ganie@novavitagroup.com
Accepted 20 September 2017


{kind=link}
{kind=link}
{kind=link}