










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.108 no.1 Pretoria Jan. 2018
http://dx.doi.org/10.7196/samj.2018.v108i1.13020
CME
N AlliI; J VaughanII; S LouwIII; E SchapkaitzIV; J MahlanguV
IMB BCh, FCPathHaem (SA); Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
IIMB BCh, FCPathHaem (SA), MMed Haem; Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
IIIMB BCh, FCPathHaem (SA), MMed Haem; Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
IVMB BCh, FCPathHaem (SA), MMed Haem; Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
VBSc, MB BCh, FCPathHaem (SA), MMed Haem, Cert Clin Haem; Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
ABSTRACT
Abnormal bleeding is a common clinical presentation in general practice, and a rational approach to this problem is therefore required. Investigation of a suspected bleeding disorder necessitates a comprehensive history, thorough physical examination and systematic laboratory work-up. Inherited bleeding disorders (IBDs) typically manifest in childhood, but may present later in life after a haemostatic challenge (such as trauma, surgery, tooth extraction). This two-part CME series is intended to provide insight to the medical practitioner on the clinical spectrum, diagnosis and management of bleeding disorders. Bleeding due to inherited disorders is the subject of discussion in part 1 (current issue), and in part 2 (forthcoming issue) the focus is on bleeding from acquired causes. Patients diagnosed with an IBD should ideally be referred to a dedicated tertiary healthcare facility, e.g. haemophilia centre, for management and follow-up.
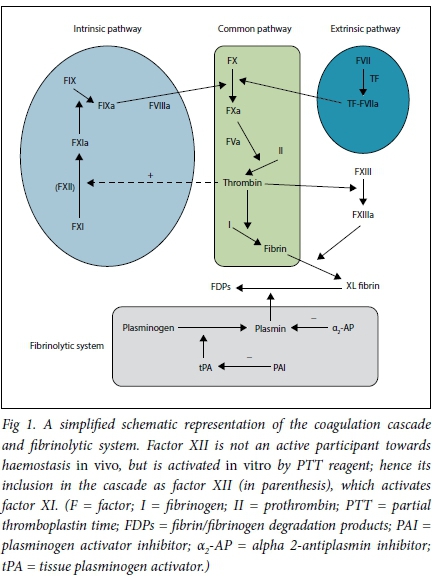
Haemostasis is a biological process that stops blood loss at the site of injury. This is accomplished in three physiological steps that occur in rapid sequence: (i) vasoconstriction; (ii) formation of a platelet plug (primary haemostasis); and (iii) stabilisation of clot through cross-linking of insoluble fibrin (secondary haemostasis). The clot is finally dissolved by the fibrinolytic system (Fig. 1).
The presence of an inherited bleeding disorder (IBD) should be suspected on presentation in early childhood or when there is a longstanding history of easy bruising, in cases of recurrent and/or severe bleeding (particularly at unusual sites) and in patients with a family history of the condition.
IBDs include a spectrum of disorders that affect clotting factors, platelets or the vessel wall, and the clinical manifestation varies depending on the underlying cause. For instance, patients with platelet and vessel wall disorders usually present with mucocutaneous bleeding (petechiae, purpura, ecchymosis, epistaxis, menorrhagia, haematuria and/or gastrointestinal tract blood loss), while clotting factor deficiencies may also cause joint and deep muscle bleeding. The most common IBD is von Willebrand disease (vWD), which affects both platelet adhesion to the vessel wall and factor VIII levels, and may therefore present with a mixed bleeding profile. The clinical history of patients should focus on the site and severity of previous episodes of bleeding, including requirements for blood transfusion, nature of the provoking injury, consequences of exposure to previous haemostatic challenges (such as surgery, tooth extraction, childbirth) and presence of a family history. A basic initial work-up includes a full blood count (FBC) with peripheral blood smear review, prothrombin time (PT)/international normalised ratio (INR) and partial thromboplastin time (PTT). If these are normal, further investigations are warranted.
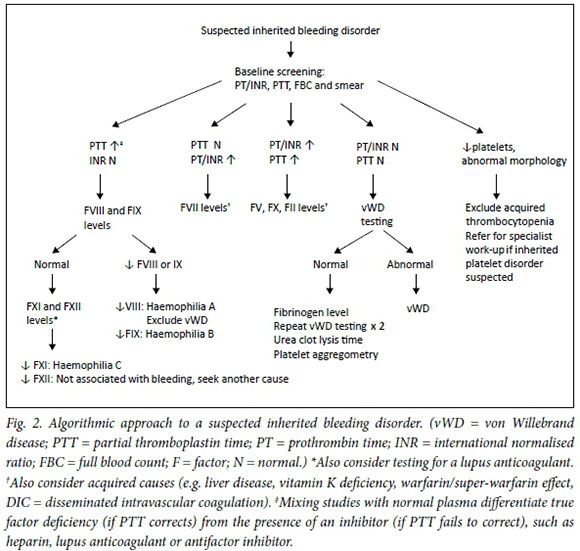
The algorithm in Fig. 2 provides a diagnostic approach for a patient presenting with a bleeding disorder. Laboratory test results of the various inherited bleeding conditions are summarised in Table 1. Each coagulation factor has a plasma half-life that determines its stability after blood sample collection. This should be kept in mind when performing laboratory tests of specific factor levels (Table 1).
To appreciate the rationale for using the various clotting tests towards achieving a diagnosis, an understanding of the coagulation cascade is necessary. Fig. 1 is a simplified schematic representation of the coagulation cascade. This is by no means complete and is intended to orientate the reader on the pathophysiology of the respective bleeding disorders. The coagulation cascade is categorised into intrinsic, extrinsic and common pathways, which are in vitro observations that assist with identifying the factor deficiency. The reagent employed to calculate the PTT activates factor XII and so measures factors in the intrinsic and common pathways. The reagent employed for the INR estimation activates factor VII and so measures the extrinsic and common pathways.
Haemophilia
Background and epidemiology
Haemophilia is an IBD due to deficiency or dysfunction of clotting factor VIII or factor IX. Clotting factor VIII is deficient in haemophilia A and factor IX is deficient in haemophilia B. Haemophilia C refers to deficiency of clotting factor XI.
The incidence is 1 in 10 000 live births in haemophilia A and 1 in 25 000 live births in haemophilia B.1 Among the inherited bleeding disorders, vWD is the most common, followed by haemophilia A and B. It is estimated that there are at least 400 000 haemophilia sufferers globally.2
Both haemophilia A and B are inherited in an X-linked fashion, with male carriers of the mutant gene afflicted with the bleeding diathesis and females being obligate carriers. Although haemophilia has a strong family history in the majority of cases, up to 30% of haemophilia cases are caused by spontaneous mutations, with no prior family history.
Clinical presentation
The clinical presentation of haemophilia takes a number of forms, including acute bleeding episodes, chronic synovitis and haemophilic arthropathy. Bleeding in haemophilia can be spontaneous or follow trauma. The hallmark of haemophilia is spontaneous intra-articular bleeding. The most commonly affected joint is the knee, followed by the ankle, elbow and hip joints.3 Bleeding into muscle, soft tissue and mucous membranes is less common, but may be organ or life threatening, e.g. intraocular bleeding, central nervous system (CNS) bleeding, and bleeds causing airway obstruction.
There is generally close correlation between the bleeding phenotype and plasma level of clotting factor, although some patients show a discrepancy between the factor level and clinical phenotype. In this regard, haemophilia is classified into severe, moderate and mild phenotypes based on the clotting factor level (Table 2).

Repeated bleeding into joints initiates a vicious cycle of synovial damage, synovial hypertrophy, neovascularisation, and repeated bleeding from the new vessels with further synovial damage. Untreated synovial damage causes harm to cartilage and articular surfaces, with consequent joint pain and ultimately joint dysfunction.
Diagnosis
Haemophilia is diagnosed by taking an appropriate bleeding history, performing a directed physical examination and confirmatory coagulation tests. A family history with an X-linked pattern of inheritance, together with intra-articular bleeding, is almost certainly associated with haemophilia.
The clinical examination may show evidence of acute joint bleeding or complications of intra-articular bleeding, such as chronic synovitis, joint deformity and limited mobility. The Haemophilia Joint Health Score and the Haemophilia Activity List are useful tools for objective assessment of joint function.
Musculoskeletal radiological assessment, where indicated, includes the affected joints, with the radiographs scored with either the Patterson or Arnold-Hilgartner scoring system. The radiological scoring systems are useful, not only in the cross-sectional assessment but also for the long-term follow-up of patients as pointers for interventions, where the scores indicate worsening joint structure.
The diagnosis of haemophilia should be confirmed with coagulation and/or genetic testing. Haemophilia typically shows a prolonged activated PTT (aPTT) with a normal INR. In the absence of neutralising antibodies, otherwise known as inhibitors, the aPTT corrects with normal plasma, indicating factor deficiency. If the prolonged aPTT does not correct, an inhibitor should be suspected. An inhibitor assay is performed using either the Bethesda or Nijmegen assay to quantify the level of the inhibitor.
Genotyping is performed to establish the nature of the mutation in the factor VIII or IX gene, as well as for inhibitor risk profiling, as certain genotypes are associated with the risk of inhibitor development. To limit costs, selective genotyping is done, i.e. inversion 22 in haemophilia A, as it is the most common genotype associated with inhibitors. Where possible, genetic testing for prenatal diagnosis should be encouraged.
Treatment
The aim of haemophilia treatment is to replace the missing clotting factor to treat or prevent bleeding. The source of clotting factor concentrate (CFC) has evolved in the last seven decades from blood and blood products to recombinant products. Both plasma-derived and recombinant CFCs are currently used. In South Africa, the CFCs are intermediate purity plasma-derived products (Haemosolvate Factor VIII and Haemosolvex Factor IX), which are fractionated locally from blood donor plasma.
Acute bleeding episodes are managed by infusing the missing clotting factor, as well as applying adjunct measures. The dose will depend on the site of bleeding. For haemarthrosis, the recommended peak factor level is 40 - 60 IU/dL for factor VIII and factor IX.1 In limb and life-threatening bleeds, the target peak level is higher - 80 - 100 IU/dL. The duration of treatment is usually 1 or 2 days, but also depends on the nature of the product used, site of bleeding and severity of bleeding.
The current standard of care in haemophilia is to prevent bleeding by prophylactic infusion of the deficient clotting factor. This dose depends on the trough level to be achieved and potential risk factors for bleeding. The frequency of infusion depends on the pharmaco-kinetic properties of the infused clotting factor. When using standard half-life products, target trough levels are 1 - 2%, while higher trough levels are achieved with extended half-life products. Standard halflife clotting factor VIII and factor IX are infused 2 - 3 times per week and 1 - 2 times per week, respectively. When using extended half-life products, the dosing interval for factor VIII and factor IX may be increased to 1 - 2 times a week and every 10 - 14 days, respectively.
With the evolving switch from plasma-derived to recombinant products and effective antiviral measures, transfusion-transmitted infections have essentially been eliminated. However, clotting factor replacement therapy is associated with development of neutralising antibodies, i.e. inhibitors, which render replacement of factor ineffective. These occur with a frequency of 20 - 30% in both plasma-derived and recombinant products. The therapeutic strategy in patients who have developed high responding inhibitors is two-fold, i.e. treatment of acute bleeding episodes and eradication of the inhibitor. For acute bleeding episodes, bypassing agents, i.e. activated prothrombin complex concentrate (aPCC) or recombinant activated factor VII (FVIIa), are shown to be safe and efficacious. The registered dose for rFVIIa is 90 μg/kg χ 3 or 270 μg/kg administered once, and that for aPCC is 50 - 100 IU/kg repeated at 12-hourly intervals. Inhibitor eradication strategies take the form of a number of protocols, including the Malmö protocol, Bonn regimen and Dutch protocol. The aim of eradication therapy is to remove the inhibitor so that patients can be treated with standard replacement therapy again. The efficacy of eradication therapy is variable, with most success rates of 80 - 100% seen in the paediatric population in the early phase of inhibitor development.
von Willebrand disease
Background
vWD is a common IBD affecting 1.3% of the population.5,6 It is caused by a quantitative (reduced amount) or qualitative (abnormal function) deficiency in von Willebrand factor (vWF), combinations of which result in the various vWD subtypes (Table 3). vWF is syn-thesised by endothelial cells and megakaryocytes and is a complex plasma protein with multiple functions, including binding to exposed sub-endothelial collagen and platelet glycoprotein Ib (GPIb) receptors, thus facilitating platelet adhesion at the site of vessel damage (primary haemostasis). vWF further binds to and prolongs the half-life of circulating coagulation factor VIII, delivering it to sites of vascular injury (secondary haemostasis). Larger vWF molecules with high molecular weight (HMW vWF multimers) facilitate better anchoring of platelets and thus thrombus formation, achieving more effective haemostasis. The complex functions of vWF can be measured by individual laboratory tests.5-7
Clinical presentation
Patients suffering from vWD usually present with mucocutaneous bleeding, including epistaxis, easy bruising, menorrhagia and excessive bleeding from minor wounds, tooth extractions and surgery. The clinical presentation is, however, very heterogeneous and the rate of spontaneous bleeding may be low, even in patients with severe vWF deficiency. There is frequently a family history of abnormal bleeding and excessive bruising, as vWD is an inherited disorder that most frequently displays an autosomal dominant pattern. Although the autosomal inheritance pattern would suggest an equal distribution between both genders, the disease is more often diagnosed in females because of female-specific haemostatic challenges, including menstruation and childbirth. vWF levels do not always correlate with bleeding symptoms and can be variable between affected family members. As factor VIII is usually only mildly reduced or normal in vWD, the manifestations in patients with haemophilia, a severe coagulation factor deficiency disorder, such as haemarthrosis and deep muscle haematomas, are rare, except in type 3 disease. Gastrointestinal bleeding also occurs and can be difficult to manage, especially in patients lacking HMW multimers. Bleeding after dental extraction is the most frequent postoperative bleeding manifestation, whereas bleeding after surgery may occur in more severely affected types 1 and 3 vWD patients. Bleeding in the postpartum period is rarely observed in type 1 vWD, as factor VIII/vWF levels correct at the end of pregnancy in mild type 1 cases. Patients with vWD types 2A, 2B and 3 usually require replacement therapy post-partum to prevent immediate or delayed haemorrhage.5-8
Diagnosis
The diagnosis of vWD, and in particular type 1, may be difficult owing to clinical heterogeneity and difficulties in standardising laboratory tests. In vWD patients, the platelet count is usually normal, except in the relatively rare type 2B subtype, in which a mild to moderate thrombocytopenia can occur. The PT is normal, whereas the aPTT may be prolonged, depending on the plasma factor VIII levels. Unfortunately, there is no single test to diagnose vWD. The standard diagnostic tests include measurement of total vWF protein (vWF antigen), a vWF activity (such as ristocetin-cofactor activity) and ristocetin-induced platelet aggregation (RIPA), which determine the ability of vWF to bind platelets. Coagulation factor VIII levels are also measured. Specialised tests, such as vWF multimers and vWF binding to platelets and factor VIII, assist with the sub-classification of vWD. Various physiological and pathological events, including pregnancy, stress and bleeding, can temporarily normalise vWF levels in patients with the condition and should be taken into account when testing for vWD. The exclusion of vWD, depending on the results of laboratory tests, is therefore only made once three normal tests separated in time are obtained. Levels of vWF vary among different ethnic groups and blood types.6,7,9
Treatment
Treatment of vWD is based on normalising vWF and factor VIII levels in cases of bleeding or before a planned intervention, such as surgery or delivery. This can be achieved with desmopressin (DDAVP) or by infusing exogenous coagulation factors in the form of a high-purity vWF concentrate or low-purity factor VIII/vWF concentrate.6 Desmopressin, an analogue of human antidiuretic hormone (ADH), releases vWF and factor VIII from endothelial cells into the circulation. vWF concentrates are used for the treatment of active bleeding or prophylaxis of bleeding with invasive procedures. There is currently no recommendation for any vWF product for routine prophylaxis to prevent recurrent, spontaneous or incidental haemorrhage.10 General measures include antifibrinolytic therapy, e.g. tranexamic acid, the combined oral contraceptive pill, avoidance of antiplatelet agents (such as aspirin) and treatment of resultant anaemia.8
Other clotting factor deficiencies
These are less common than haemophilia A and B, and include deficiencies of factors V, VII, X, XI, XIII and fibrinogen, and protease inhibitors. The mode of inheritance is usually autosomal recessive. The expected results of diagnostic tests are summarised in Table 1.
Fibrinogen abnormalities
Afibrinogenaemia/hypofibrinogenaemia
This bleeding disorder is rare and surprisingly less severe than haemophilia, where 20% of patients develop haemarthrosis. Patients generally present with bleeding at mucosal sites, e.g. menorrhagia and umbilical stump bleeding in neonates. Although severe bleeding is uncommon, intracranial haemorrhage in childhood is the leading cause of death.11 First-trimester abortion is common in females with afibrinogenaemia.
Dysfibrinogenaemias are qualitative defects that may be associated with bleeding or thrombosis, depending on the site of mutation. Patients may present with defective wound healing and recurrent miscarriages. The mode of inheritance is autosomal dominant.
Treatment for bleeding due to fibrinogen abnormalities is on-demand fibrinogen replacement with fresh frozen plasma, cryoprecipitate or fibrinogen concentrate during bleeding episodes. For frequent or life-threatening bleeding episodes, prophylactic fibrinogen replacement every 7 - 14 days should be considered.
Prothrombin, and factors V, VII and X
Deficiencies of any of the above-mentioned factors are rare, of which factor VII deficiency is the most common autosomal recessive coagulation disorder (frequency 1:500 000) and prothrombin deficiency the rarest (1:2 000 000). Severity of bleeding episodes is variable and may occur at various sites, including mucosal surfaces, intra-articular sites, gastrointestinal tract and CNS. Spontaneous haemorrhage is uncommon.
Combined factor V and factor VIII deficiency has been described in some families,121 and should be treated with replacement of factor in various preparations, depending on availability, i.e. fresh frozen plasma, PCC, factor VII concentrate or recombinant factor preparations.
Factor XI deficiency (haemophilia C)
Factor XI deficiency occurs at an overall frequency of 1:1 000 000, but is more common in Ashkenazi Jews with homozygous and heterozygous frequencies of 0.3% and 8.0%, respectively. The extent of bleeding manifestations is variable, but usually occurs after surgery or trauma. Spontaneous bleeding is uncommon. Treatment includes factor XI replacement (fresh frozen plasma, factor XI concentrate) or antifibrinolytic agents.
Kindred with combined factor XI and factor IX deficiency have been described.13 Factor XI deficiency may be encountered in Gaucher's disease and Noonan's syndrome.14,15
Factor XIII deficiency
Factor XIII stabilises the clot by forming intermolecular covalent bonds between fibrin molecules. In deficient states, bleeding may occur with varying severity at different sites, including mucosal surfaces, soft tissue, intra-articular, intraperitoneal and CNS sites. Severe bleeding episodes in neonates are common, viz. umbilical stump (80%) and CNS bleeding (up to 30%). Deficiency of factor XIII should be suspected when there is delayed wound healing or a bleeding disorder in the presence of a normal PTT, PT/INR, bleeding time and platelet count.
As factor XIII has a long half-life, prophylactic replacement therapy every 4 weeks with factor XIII concentrate or cryoprecipitate is effective as a preventive strategy.
Protease inhibitors
Protease inhibitors include alpha 2-antiplasmin, plasminogen activator inhibitor-1 and alpha 1-antitrypsin.
Alpha 2-antiplasmin deficiency is inherited as a rare autosomal recessive disorder. The homozygous form causes severe bleeding episodes. Suspicion of the condition is aroused when the fibrinogen level is decreased and the urea clot lysis time is shortened. Confirmation of the diagnosis is obtained through fluorometric or chromogenic assays.
Vitamin K-dependent coagulation factor deficiency
The vitamin K-dependent factors II, VII, IX and X require gamma-carboxylation of glutamic acid residues to enable binding to Ca++. Vitamin K-dependent coagulation factor deficiency is caused by deficiency of enzymes responsible for gamma-carboxylation. Patients present with bleeding of varying degrees of severity and may have associated skeletal abnormalities.
The mode of treatment is parenteral vitamin K administration, and fresh frozen plasma or PCC if response is suboptimal.
Inherited platelet function disorders
Platelet structure and function
Platelets are small (2 pm) anuclear cells produced by megakaryocytes in the bone marrow. In the resting state, platelets have a smooth discoid shape and contain cytoplasmic organelles, cytoskeletal elements, platelet-specific alpha and dense granules and an invaginating open-cannalicular phospholipid membrane. In the outer plasma membrane there are numerous receptors for ligands, such as fibrinogen, collagen, thrombin and vWF for primary haemostasis.
Platelets promote primary haemostasis by four mechanisms16The initial step is platelet adhesion. Following damage to the vessel wall, platelets undergo rapid activation by thrombin. vWF is a large, multimeric protein secreted from endothelial cells. At high shear rates, the platelet GP1b undergoes a conformational change, which promotes platelet adhesion to vWF. Platelet adhesion stimulates intracellular signalling, leading to granule release of alpha granules (platelet factor 4, thrombospondin, platelet-derived growth factor, factor V, fibrinogen, vWF, beta-thromboglobulin) and dense granules (calcium, adenosine diphosphate (ADP), serotonin). ADP release causes a conformational change of the fibrinogen receptor, GPIIb/ IIIa. Fibrinogen forms a bridge between adjacent platelets, resulting in platelet aggregation and the formation of a primary platelet plug. Activated platelets also expose negatively charged phospholipids on the surface membrane, which provides a procoagulant surface for the formation of a secondary stable clot. Platelet disorders can be classified according to disorders of platelet function.
Clinical presentation
Inherited platelet disorders are a rare, wide spectrum of disorders characterised by a variable bleeding pattern. This usually presents as prolonged bleeding after haemostatic challenges or in childhood, with spontaneous mucocutaneous bleeding from gastrointestinal and genitourinary sites.17
Laboratory investigations
A careful medical, drug and family bleeding history should be taken and a physical examination should be performed before requesting laboratory investigations for platelet disorders (Table 4).18 The traditional screening tests include a FBC, peripheral blood smear review and bleeding time/platelet function assay. The normal range for the platelet count is 150 - 400 χ 109/L. Qualitative platelet disorders can be distinguished by morphological features. The bleeding time test has largely been replaced with automated platelet function assays as a screening test to evaluate platelet function because of the poor reproducibility and wide variability of bleeding time tests.19

Definitive diagnosis requires specific tests, which are available in specialist laboratories.20 Platelet aggregation studies measure the ability of agonists to cause in vitro platelet activation and binding. Flow cytometry is used to diagnose deficiencies of platelet glycoproteins with fluorescently labelled antibodies. Lastly, electron microscopy (EM) is useful for the evaluation of the ultrastructure of platelets.
The more common inherited platelet disorders
• Glanzmann thrombasthenia is a severe, autosomal recessive bleeding disorder caused by a deficiency of GPIIb/IIIa, the receptor responsible for platelet aggregation. The diagnosis is made on platelet aggregation or flow cytometry studies. No aggregation response to ADP agonists, collagen, adrenaline and arachidonic acid occurs.
• Bernard-Soulier syndrome is an autosomal recessive deficiency of the platelet GP1b/IX/V receptor, which is responsible for platelet adhesion. Patients with Bernard-Soulier syndrome also have a moderate thrombocytopenia, with large platelets. The diagnosis can be confirmed on platelet aggregation or flow cytometry studies. There is a normal aggregation response to ADP agonists, collagen, adrenaline and arachidonic acid, but aggregation is absent with the addition of ristocetin.
• Secretory disorders occur secondary to deficiencies of alpha and/ or dense granules or defects in signal transduction. Dense granule storage pool disorders (SPDs) have decreased aggregation with ADP, adrenaline and collagen and often occur in association with other hereditary disorders. EM usually shows decreased dense granules. Gray platelet syndrome is an autosomal dominant alpha SPD characterised by mild bleeding symptoms. Platelets appear grey due to decreased alpha granules.
• May-Hegglin anomaly is a group of autosomal dominant thrombocytopenias consisting of May-Hegglin anomaly and Sebastian, Fechtner and Epstein syndromes and is characterised by large platelets and thrombocytopenia (20 - 130 χ 109/L). It presents as a mild bleeding disorder. There is a normal aggregation response. EM confirms the diagnosis.
• Scott syndrome is a rare congenital disorder due to defective platelet membrane flip-flop action, with decreased exposure of procoagulant platelet membrane surfaces.
Management
The cornerstone of management is supportive care. Antifibrinolytics, e.g. tranexamic acid, are an important adjuvant. Specific management depends on the particular type of disorder, as well as the severity of the bleeding. Desmopressin has been shown to be effective in SPDs and mild platelet function disorders, and may be of benefit in Bernard-Soulier syndrome. Platelet transfusions and rFVIIa are used in cases of severe/life-threatening bleeding.
In summary, inherited platelet disorders are a rare group of disorders characterised by mild to severe mucocutaneous bleeding. A careful bleeding history should be taken before referring patients to specialist laboratories for investigations for platelet disorders. The latter should be managed at specialist haemophilia centres.21
Vessel wall defects
Vessel wall abnormalities are among the IBDs that are associated with normal screening tests. They include hereditary haemorrhagic telangiectasia (HHT) (an autosomal dominant structural vascular abnormality) and hereditary connective tissue disorders (such as Ehlers-Danlos syndrome and osteogenesis imperfecta). Of these, HHT is the most common, with an incidence of ~1:5 000 - 10 000. It presents in the 2nd or 3rd decade of life with telangiectatic lesions (0.5 - 1 mm red lesions that blanche with pressure), which are typically seen on the lips, oral mucosa and finger tips. It is typified by mucosal bleeding (commonly epistaxis or gastrointestinal blood loss), with consequent iron deficiency. Catastrophic bleeding is uncommon. HHT may have associated arteriovenous malformations/fistulae of solid organs (particularly the lung, liver and CNS). Arteriovenous fistulae may be associated with significant complications, such as embolic phenomena, high-output cardiac failure and portal hypertension. Management includes iron supplementation, laser ablation and, in the case of arteriovenous fistulae, embolisation.
Conclusion
Abnormal bleeding is commonly encountered in general practice, and a rational approach to this problem is therefore required. Investigation of abnormal bleeding requires a comprehensive history, thorough physical examination and systematic laboratory work-up. Patients diagnosed with an IBD should be referred to a tertiary healthcare facility, such as a haemophilia centre for long-term management.
Acknowledgements. Mr C Moodly for reviewing the technical aspects of laboratory tests.
Author contributions. Selected sub-sections prepared by respective authors listed. NA and JV: review of manuscript; and NA: co-ordination and editing of information.
Funding. None.
Conflicts of interest. None.
References
1 . Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia 2013;19(1):e1-e47. https://dx.doi.org/10.1111/j.1365-2516.2012.02909.x [ Links ]
2. Stonebraker JS, Bolton-Maggs PH, Soucie JM, Walker I, Brooker M. A study of variations in the reported haemophilia: A prevalence around the world. Haemophilia 2010;16(1):20-32. https://dx.doi.org/10.1111/j.1365-2516.2011.02588.x [ Links ]
3. Mahlangu JN, Gilham A. Guideline for haemophilia treatment in South Africa. S Afr Med J 2008;98(2):126-140. [ Links ]
4. White GC, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost 2001;85(3):560. [ Links ]
5. Echahdi H, El Hasbaoui B, El Khorassani M, et al Von Willebrand's disease: Case report and review of literature. Pan Afr Med J 2017;27:147. https://dx.doi.org/10.11604/pamj.2017.27.147.12248 [ Links ]
6. Leebeek FW, Eikenboom JC. Von Willebrand's Disease. N Engl J Med 2017;376(7):701-702. https://dx.doi.org/10.1056/NEJMc1616060 [ Links ]
7. Bowman ML, James PD. Controversies in the diagnosis of type 1 von Willebrand disease. Int J Lab Hematol 2017;39(Suppl 1):61-68. https://dx.doi.org/10.1111/ijlh.12653 [ Links ]
8. Castaman G, Linari S. Diagnosis and treatment of von Willebrand disease and rare bleeding disorders. Clin Med 2017;6(4):E45. https://dx.doi.org/10.3390/jcm6040045. [ Links ]
9. Montgomery RR, Flood VH. What have we learned from large population studies of von Willebrand disease? Hematol Am Soc Hematol Educ Program 2016;2016(1):670-677. https://dx.doi.org/10.1182/asheducation-2016.1.670 [ Links ]
10. Neff AT. Current controversies in the diagnosis and management of von Willebrand disease. Ther Adv Hematol 2015;6(4):209-216. https://doi.org/10.1177/2040620715587879 [ Links ]
11. Montgomery R, Natelson SE. Afibrinogenemia with intracerebral hematoma: Report of a successfully treated case. Am J Dis Child 1977;131:555-556. [ Links ]
12. Seligsohn U, Zivelin A, Zwang E. Combined factor V and factor VIII deficiency among non-Ashkenazi Jews. N Engl J Med 1982;307(19):1191-1195. https://dx.doi.org/10.1056/NEJM198211043071907 [ Links ]
13. Soff GA, Levin J, Bell WR. Familial multiple coagulation deficiencies. Combined factor VIII, IX and XI deficiency and combined IX and XI deficiency: Two previously uncharacterized familial multiple factor deficiency syndromes. Sem Thromb Hemost 1981;7(2):149-169. https://dx.doi.org/10.1055/s-2007-1005074 [ Links ]
14. Berrebi A, Malnick SDH, Vorst EJ, et al High incidence of factor XI deficiency in Gaucher's disease. Am J Hematol 1992;40(2):153. https://dx.doi.org/10.1002/ajh.2830400215 [ Links ]
15. Kitchens CS, Alexander JA. Partial deficiency of coagulation factor XI as a newly diagnosed feature of Noonan syndrome. J Pediatr 1983;102(2):224-227. https://doi.org/10.1016/s0022-3476(83)80525-3 [ Links ]
16. Gresele P; Subcommittee on Platelet Physiology of the International Society on Thrombosis and Hemostasis. Diagnosis of inherited platelet function disorders: Guidance from the SSC of the ISTH. J Thromb Haemost 2015;13(2):314-322. https://doi.org/10.1111/jth.12792 [ Links ]
17. Kenny D, Mezzano D, Mumford AD, et al. Diagnosis of suspected inherited platelet function disorders: Results of a worldwide survey. J Thromb Haemost 2014;12(9):1562-1592. https://dx.doi.org/10.1111/jth.12650 [ Links ]
18. Israels SJ, El-Ekiaby M, Quiroga T, Mezzano D. Inherited disorders of platelet function and challenges to diagnosis of mucocutaneous bleeding. Haemophilia 2010;16:152-159. https://dx.doi.org/10.1111/j.1365-2516.2010.02314.x [ Links ]
19. Hayward CP, Rao AK, Cattaneo M. Congenital platelet disorders: Overview of their mechanisms, diagnostic evaluation and treatment. Haemophilia 2006;12(3):128-136. https://dx.doi.org/10.1111/j.1365-2516.2006.01270.x [ Links ]
20. Bolton-Maggs PH, Chalmers EA, Collins PW, et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol 2006;135(5):603-633. https://dx.doi.org/10.1111/j.1365-2141.2006.06343.x [ Links ]
21. Geddis AE. Inherited thrombocytopenias: An approach to diagnosis and management. Int J Lab Hematol 2012;35(1):14-25. https://dx.doi.org/10.1111/j.1751-553X.2012.01454.x [ Links ]
22. Kumar N, Garg N, Khunger M, Gupta A. Optimal management of hereditary hemorrhagic telangiectasia. J Blood Med 2014;5:191-206. https://doi.org/10.2147/JBM.S45295 [ Links ]
 Correspondence:
Correspondence:
N Alli
nazeer.alli@nhls.ac.za
Accepted 24 November 2017


{kind=link}
{kind=link}