










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.107 no.5 Pretoria Mai. 2017
http://dx.doi.org/10.7196/samj.2017.v107i5.9461
RESEARCH
Osteogenesis imperfecta type 3 in South Africa: Causative mutations in FKBP10
A VorsterI; P BeightonII; M ChettyIII; Y GanieIV, V; B HendersonVI; E HoneyVII; P MaréVIII; D ThompsonIX; K FieggenX; D ViljoenXI; R RamesarXII
IMSc; MRC Human Genetics Research Unit, Division of Human Genetics, Institute for Infectious Diseases and Molecular Medicine, Department of Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
IIMD, PhD, FRCP, FRSSA; MRC Human Genetics Research Unit, Division of Human Genetics, Institute for Infectious Diseases and Molecular Medicine, Department of Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
IIIBChD, MChD, PhD; Department of Oral and Molecular Biology, Faculty of Dentistry, University of the Western Cape, Cape Town, South Africa
IVMB ChB, FCP (Paeds);Department of Paediatrics and Child Health, School of Clinical Medicine, College of Health Sciences, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
VMB ChB, FCP (Paeds); Division of Paediatric Endocrinology, Inkosi Albert Luthuli Central Hospital, Durban, South Africa
VIMB ChB, DCH, MMed; Division of Clinical Genetics, Department of Neurology, Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
VIIMB ChB, MMed (Paed); Department of Genetics, Faculty of Health Sciences, University of Pretoria, South Africa
VIIIMB ChB, FCOrth (SA); Paediatric Orthopaedic Unit, Department of Orthopaedic Surgery, Grey's Hospital, Pietermaritzburg, and School of Clinical Medicine, College of Health Sciences, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
IXMB ChB, FRCS (Glasg); Paediatric Orthopaedic Unit, Department of Orthopaedic Surgery, Grey's Hospital, Pietermaritzburg, and School of Clinical Medicine, College of Health Sciences, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
XMB ChB, FCPaed, Cert Med Genet; Department of Medicine, Faculty of Health Sciences, University of Cape Town, South Africa
XIMB ChB, FCP; Foundation for Alcohol Related Research (FARR), Rondebosch, Cape Town, South Africa
XIIMSc, PhD, Exec MBA; MRC Human Genetics Research Unit, Division of Human Genetics, Institute for Infectious Diseases and Molecular Medicine, Department of Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
BACKGROUND. A relatively high frequency of autosomal recessively inherited osteogenesis imperfecta (OI) type 3 (OI-3) is present in the indigenous black southern African population. Affected persons may be severely handicapped as a result of frequent fractures, progressive deformity of the tubular bones and spinal malalignment.
OBJECTIVE. To delineate the molecular basis for the condition.
METHODS. Molecular investigations were performed on 91 affected persons from seven diverse ethnolinguistic groups in this population.
RESULTS. Following polymerase chain reaction amplification and direct cycle sequencing, FKBP10 mutations were identified in 45.1% (41/91) OI-3-affected persons. The homozygous FKBP10 c.831dupC frameshift mutation was confirmed in 35 affected individuals in the study cohort. Haplotype analysis suggests that this mutation is identical among these OI-3-affected persons by descent, thereby confirming that they had a common ancestor. Compound heterozygosity of this founder mutation was observed, in combination with three different deleterious FKBP10 mutations, in six additional persons in the cohort. Four of these individuals had the c.831delC mutation.
CONCLUSION. The burden of the disorder, both in frequency and severity, warrants the establishment of a dedicated service for molecular diagnostic confirmation and genetic management of persons and families with OI in southern Africa.
Osteogenesis imperfecta (OI) is a heterogeneous group of disorders in which skeletal fragility and frequent fractures are the major features. The classification and nosology of OI has been through a number of revisions reflecting the discovery of new subtypes, initially as clinical entities and subsequently defined by their molecular aetiology. With increasing knowledge and access to molecular genetic testing, it has become evident that there is substantial phenotypic overlap with mutations in the same gene. This prompted the International Nomenclature Committee for Constitutional Disorders of the Skeleton in 2015 to propose a return to a clinical classification of five subtypes, as proposed by Van Dijk and Sillence[1] and illustrated in Table 1.

In clinical medicine, the most common form of the condition is OI type 1 (OI-1). This autosomal dominant (AD) disorder varies in severity, but it is usually milder than other types of OI. The other well-known condition, OI type 2 (OI-2), causes stillbirth or death in the neonate. In this disorder, the fracturing tendency leads to a characteristic radiological appearance. A third form of OI, termed OI-3, has become the focus of medical and scientific interest. This severe condition, which is genetically heterogenous, has the descriptive title 'progressively deforming OI'. Genetic transmission of OI-3 can be AD or autosomal recessive (AR), but in the South African (SA) population it is most frequently AR.[2] OI-4 is typically of moderate severity, whereas OI-5 is characterised by the radiological appearance of calcification of interosseous membranes.
A severe AR form of OI-3 is present in a relatively high frequency in several indigenous black populations of southern Africa.[3] This condition is characterised by numerous fractures, gross deformity of tubular bones, spinal malalignment and marked impairment of growth. Multiple fractures may be present at birth, but the specific radiological appearances in the newborn have not yet been documented. Physical handicap is severe and affected children often become wheelchair bound. Death before adulthood is frequent. Congenital contractures are present in some affected persons; this complication has the eponymic designation Bruck syndrome.[4]
The phenotypic manifestations and orthopaedic complications in 21 indigenous black African children with AR OI-3 were tabu-lated'51 and the radiological manifestations in this age group were subsequently documented in detail.[6] The high prevalence of OI-3 in the black population was emphasised when a minimum population frequency of 0.6 per 100 000 was established on a basis of 26 affected persons in 15 families living in the Johannesburg region of SA.[3] In contrast, only five affected individuals in this black African community had AD OI-1, which is common worldwide. An additional 42 children of Shona and Ndebele heritage with OI-3 were identified in Zimbabwe, and by the end of 1987, a total of 75 affected children in 38 families had been documented.[7] The clinical and radiological phenotype was similar in each population, and it was proposed that the determinant mutation or mutations may be common in the indigenous black communities in southern Africa. Whether the geographical and demographic distribution of this disorder is the result of selective biological advantage or a founder effect, or both, is a matter for speculation.[8]
Although OI-3 is rare, studies in affected persons from different parts of the world have revealed causative mutations in several genes: CRTAP, P3H1, FKBP10, PPIB, SERPINH1, SERPINF1, BMP1, SP7, WNT1, TMEM38B, PLOD2 and CREB3L1.[9] Mutations in FKBP10, which encodes FKBP65, have been reported in affected persons of Turkish, Mexican-American,[10] German,[11] Saudi Arabian,[12] indigenous black South African,[13] Indonesian,[14] Egyptian,[15] Italian,[16] Palestinian,[17] Lebanese, Sudanese,[18] Samoan, North American[17,19] and Chinese ancestry.[20] In this context, homozygosity for a specific nucleotide insertion, NM_021939.3:c.831dupC, in the FKBP10 gene has been demonstrated in OI-3 patient populations indigenous to Europe,[10-13] Asia,[21] Africa[13,18] and the Middle East.[12]
Objective
In view of the clinical importance of OI-3 in SA and the fact that several different mutations have been shown to underlie this condition in other populations, it was imperative that the molecular basis of OI-3 in SA should be elucidated.
Methods
Ascertainment
The OI-3 phenotype in SA black children was first recognised as an autonomous AR entity by author PB at the Orthopaedic Clinic, Chris Hani Baragwanath Hospital, Johannesburg, in 1970. Thereafter, numerous affected children of indigenous black African ancestry were seen and documented in outreach clinics and institutional facilities for the physically handicapped throughout southern Africa. From 1972 onwards, data and radiographs were archived in the Division of Human Genetics, University of Cape Town (UCT). Biological material was obtained from affected children with parental permission and was retained for future studies. This investigation received clearance from the Faculty of Health Human Research Ethics committee at UCT (ref. nos 203/2013 and 192/2014).
Prior to the current project, contact was made with medical colleagues in other centres in SA, and they were offered molecular genetic screening for mutations in their patients with OI-3 by the Division of Human Genetics at UCT. The clinicians caring for affected persons with OI-3 and their families submitted blood or saliva together with signed consent and permission forms in accordance with standard ethical protocols. The clinical diagnosis of OI-3 was confirmed by the medical attendant prior to molecular investigation in every instance. The molecular findings were transmitted to the referring clinicians for the purpose of further medical and genetic management. Specimens from a total of 91 persons of indigenous black African ancestry were investigated, the majority of whom were children. The linguistic subgrouping of these persons reflected the geographical distribution of their communities and the whereabouts of the referring clinicians and hospital services.
Molecular laboratory investigations
Genomic DNA (100 ng) was amplified by standard polymerase chain reaction (PCR) methods for the exon 5 coding region of FKBP10 in 91 OI-3-affected persons. DNA sequencing was undertaken on the ABI3130x/ Genetic Analyser using the BigDye v3 terminator chemistry (ThermoFisher Scientific, USA) to investigate the region of FKBP10 in which the c.831dupC duplication had previously been identified.[10,13] Similarly, DNA from two affected persons with a heterozygous c.831dupC mutation was PCR amplified and sequenced for the remaining coding regions of FKBP10 in an attempt to identify both disease-causing mutations. Owing to financial constraints it was not possible to sequence FKBP10 in the remaining study cohort.
Two fluorescently labelled multi-allelic markers, rs59064727 and rs56965181, were typed by PCR amplification, subsequent capillary electrophoresis and fragment analysis on the ABI3130x/ Genetic Analyser. Allele frequencies were determined for 47 apparently healthy persons from the background indigenous black African population group. Genotyping was completed for 86 of the 91 OI-3-affected persons for whom genomic DNA was available. The resulting genotypes were employed to estimate the extent to which OI-3-affected persons carrying the c.831dupC causative mutation shared an 85 848-base pair genomic segment, or haplotype, in the vicinity of the FKBP10 gene. The statistical software package PHASE version 2.1.1 (Stephens Lab, University of Chicago, USA),[22] was used to estimate haplotype frequencies in the indigenous black African population and to infer haplotypes in the OI-3-affected persons. Oligonucleotide sequences employed for amplification of multi-allelic markers and the FKBP10 coding regions are presented in Table 2.
Results
Molecular pathology
FKBP10 mutations were identified in 45.1% (41/91) of the OI-3-affected persons. Details of mobility status, z-scores, centiles, age, gender, linguistic group and investigation centre are set out in Table 3.
Two frameshift and two nonsense mutations were identified as the disease-causing DNA variations in OI-3-affected persons in the study cohort. The previously reported frameshift mutation, c.831dupC p.(Gly278Argfs95*),[10] was identified, homozygous, in 38.5% (35/91) of individuals in the study cohort. The c.831dupC mutation is predicted to substitute a glycine with an arginine at amino acid residue 278 of FKBP65 with the introduction of a premature termination codon 95 residues from this substitution site. In combination with this common mutation, a novel frameshift mutation, c.831delC p.(Gly278Alafs20*), was identified in four compound heterozygous OI-3-affected persons from the study cohort (Fig. 1). This mutation, unlike the c.831dupC variant, is predicted to substitute the glycine 278 residue with an alanine, with subsequent introduction of a premature termination codon 20 residues from the substitution site. The novel c.343C>T p.(Arg115*) and c.1621C>T p.(Gln541*) nonsense mutations are predicted to truncate FKBP65 by 467 and 41 amino acid residues, respectively.
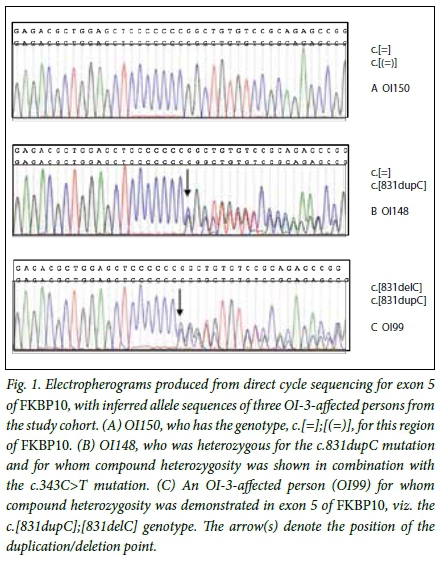
In a further 50 affected persons, no mutations were identified in the FKBP10 regions that were studied.
Haplotype analysis
Haplotypes were constructed to establish whether the high frequency of the c.831dupC FKBP10 mutation, present in the OI-3-affected population, is the result of a single mutational event in a common ancestor, or whether it is the result of a mutational hotspot. Nineteen likely haplotypes were inferred with the PHASE version 2.1.1 software in 86 of the 91 OI-3-affected individuals in this study. Every person harbouring the c.831dupC mutation and for whom genotyping was completed (n=37) was predicted to share the same disease-associated haplotype (3-1) around FKBP10 for rs59064727 (allele 3) and rs56965181 (allele 1). Persons homozygous for the c.831dupC mutation were also homozygous for these alleles. Based on the observed allele frequencies for rs59064727 and rs56965181 in 47 unaffected persons from the background indigenous black African population, the frequency of the disease-associated haplotype (3-1) was estimated at 4%. The c.831dupC mutation is therefore not present on a common haplotype in the background population, suggesting that the mutation is identical among the patient population, by descent. It is likely that the initial causative mutation occurred in the indigenous black African population group before the divergence of the linguistic groups that are shown in the OI-3 study cohort (Table 3). The c.831delC mutation-associated haplotype in the four compound heterozygous individuals with the genotype c.[831dupC];[831delC] was 3-7 for rs59064727-rs56965181.
Clinical manifestations
The clinical features in the individuals with OI-3 were generally much more severe than those of the common OI-1. There were no obvious differences in the phenotypic manifestations in the mutation-positive and mutation-negative persons in this investigation. The affected persons experienced frequent fractures in early childhood and many developed limb deformities. Progressive spinal malalignment was frequent. Growth was impaired and reduced stature was usual. It is significant that all the patients in whom height or length was measured and z-scores calculated fell below the 5th centile (Table 3). Three individuals had congenital contractures in the limbs that were consistent with a diagnosis of the complication termed Bruck syndrome.
The radiological phenotype reflected the sequelae of limb fracturing and deformation, together with spinal malalignment. The characteristic wormian bones were present in the cranial sutures. Elongation of the vertebral pedicles and protrusio acetabulae in the pelvis were useful diagnostic indicators.[6]
In all affected individuals the sclerae were white, and a blue tinge that was sometimes present in infancy was within normal limits. Intellect was normal and there were no relevant visceral complications. The 41 homozygotes and compound heterozygotes had no clinically evident dentinogenesis imperfecta. In the 50 persons in whom no mutation in FKBP10 was detected, dentinogenesis imperfecta was variable.
Until recently, the natural history of OI-3 in southern Africa was progressive limb and spinal malalignment, with impairment of locomotion in early childhood. Walking aids such as sticks and crutches often became necessary, spinal compression sometimes supervened, and a wheelchair existence was followed by death from cardiorespiratory impairment before adulthood. With the availability of bisphosphonate therapy, the incidence of complications has decreased and long-term prognosis has improved.[23] This has changed the age relationships of the phenotype, thereby complicating the establishment of genotype-phenotype correlations.
The molecular status of each affected individual is listed in Table 3, together with their linguistic and geographical group. In clinical medicine, number of fractures, height and the degree of restriction of physical mobility are used as an expression of severity of the condition in individuals with OI. It is relevant that these complications are influenced by numerous exogenous factors such as diet, general health and social circumstances. As the project was undertaken in several separate academic centres, observational variation introduces additional complexity. For all these reasons, statistical analysis would have been meaningless and was not undertaken. Nevertheless, we consider that the phenotypic information that is presented provides a useful indication of the clinical severity in the affected persons. It can be concluded that there were no obvious discrepancies in the correlation of the phenotype with any of the specific mutations recognised in the regions of FKBP10 that were investigated.
Discussion
Although the syndromic identity of the SA form of OI-3 was recognised more than 40 years ago, until recently the nature of the basic defect has remained enigmatic. The observation of a specific mutation in the FKBP10 gene in an affected SA boy[13] provided the key to elucidation of the molecular pathogenesis, which we have now confirmed in a large series of persons with the condition in this country. Our molecular investigations were restricted to a small region of the FKBP10 gene, and it is relevant that mutations were identified in only 45.1% (41/91) of the affected individuals. In an additional 50 persons with an identical phenotype in the same population group, no mutation was identified in molecular regions that were studied, and it seems very probable that further genetic heterogeneity remains to be recognised in FKBP10 and possibly other genes in this population. The search for the missing mutation or mutations is continuing.
The observation of an expansion (c.831dupC) and contraction (c.831delC) of the seven cytosine mononucleotide repeat (Fig. 1) in OI-3-affected persons from this study cohort supports the notion that this region of the FKBP10 gene is prone to mutation.[19] This increased mutability may in part contribute to the presence of the c.831dupC mutation across geographically diverse population groups worldwide. Nevertheless, the evidence suggests that the comparatively high incidence of the mutation in southern African populations is the result of a common ancestral founder effect.
Two decades ago when relevant biochemical and molecular technology became available, involvement of the COL1A1 and COL1A2 genes was excluded and it was suggested that in this AR form of OI, genes that controlled the processing of type I collagen may have been involved.[24] It has now been established that the biomolecular mechanism in some forms of AR OI-3 involves post-translational modification of collagen. In particular, 3-prolyl hydroxylation of fibrillar cartilage is controlled by a complex of the proteins encoded by the CRTAP and P3H1 genes. Mutations in these genes result in over-modification of collagen.[25-27] In the forms of AR OI-3 in which collagen is not over-modified, mutations were identified in the FKBP10 and SERPINH1 genes, which chaperone the collagen heterotrimer through the endoplasmic reticulum.[10,28]
The association of congenital joint contractures and OI-3 was initially documented in five children from families of different linguistic groups in the black population of southern Africa (Tswana, Pedi and Shona) by Viljoen et al.[29] These authors used the eponymous designation Bruck syndrome, based on historical precedents.[4] The orthopaedic aspects of additional affected children (Venda) were reviewed by Mokete et al.,[30] and DNA was provided for a large-scale international investigation undertaken by Kelley et al.[13] This study involved molecular screening of material from 47 affected individuals with OI in whom the COL1A1, COL1A1, CRTAP and P3H1 genes were normal.[13] In four families a total of five affected individuals had mutations in the FKBP10 gene. Sequence analysis carried out in an affected SA boy from a consanguineous Venda family revealed homozygosity for a single nucleotide insertion in exon 5, which resulted in a frame shift and premature stop codon p.(Gly278Argfs*95). His sister had the typical manifestations of OI-3 without any contractures, but molecular studies were not undertaken in this girl. Homozygosity for the same mutation was also identified in an affected daughter from a consanguineous Turkish family.[10] A second unrelated indigenous black SA girl with Bruck syndrome from a non-consanguineous Tswana family was shown to have compound heterozygosity involving this mutation plus another single nucleotide insertion in exon 8.[13] These findings by Kelley et al.[13] provided impetus for the project that forms the subject of this article.
Conclusion
Now that specific mutations in FKBP10 have been recognised, it would be appropriate to establish a dedicated genetic service for affected families in southern Africa. This approach could include molecular diagnostic confirmation, carrier detection, cascade screening, antenatal diagnosis and preimplantation genetic diagnosis. In clinical practice in southern Africa, it would be reasonable to consider the diagnosis of OI-3 in any person in the indigenous black African community with the generic features of OI, especially if the manifestations are severe. The white colour of the sclerae and the AR mode of transmission in the family would be further diagnostic factors in OI-3. In view of the large number of affected children in this country and the severity of the condition, this service would be cost-effective in terms of both monetary and humanitarian perspectives.
Acknowledgements. We thank Prof. Lizette van Rensburg of the Department of Human Genetics at the University of Pretoria for extraction of genomic DNA from whole blood of persons residing in that geographical region. In accordance with UCT's policy, some dentally related data in this article have been reported in a PhD thesis submitted by Dr Manogari Chetty.
Author contributions. AV undertook the molecular investigations and wrote the relevant part of the manuscript; PB delineated the disorder, and wrote the general and clinical component of the manuscript; MC travelled extensively in SA in order to examine the cranial facial components of the condition, assisted in the organisation, collection and transmission of biological specimens, and contributed to the manuscript; YG, BH, EH, PM, DT, KF and DV confirmed the diagnosis in the affected persons in their care, provided biological specimens and contributed to the manuscript; and RR (head of department) supervised and facilitated the molecular investigation.
Funding. Support was received by PB from the South African Medical Research Council and the National Research Foundation and used to support clinical and molecular aspects of the investigation.
Conflicts of interest. None.
References
1. Van Dijk FS, Sillence DO. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 2014;164A(6):1470-1481. http://dx.doi.org/10.1002/ajmg.a.36545 [ Links ]
2. Sillence DO, Barlow KK, Cole WG, Dietrich S, Garber AP, Rimoin DL. Osteogenesis imperfecta type III: Delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet 1986;23(3):821-832. http://dx.doi.org/10.1002/ajmg.1320230309 [ Links ]
3. Beighton P, Versfeld GA. On the paradoxically high relative prevalence of osteogenesis imperfecta type III in the black population of South Africa. Clin Genet 1985;27(4):398-401. http://dx.doi.org/10.1111/j.1399-0004.1985.tb02282.x [ Links ]
4. Bruck A. Multiple fractures associated with joint ankylosis and muscle atrophy. Dtsch Med Wochenschr 1897;23(10):152-155. [ Links ]
5. Beighton P, Spranger J, Versfeld G. Skeletal complications in osteogenesis imperfecta: A review of 153 South African patients. S Afr Med J 1983;64(15):565-568. [ Links ]
6. Versfeld GA, Beighton PH, Katz K, Solomon A. Costovertebral anomalies in osteogenesis imperfecta. J Bone Joint Surg Br 1985;67(4):602-604. [ Links ]
7. Viljoen D, Beighton P. Osteogenesis imperfecta type III: An ancient mutation in Africa? Am J Med Genet 1987;27(4):907-912. http://dx.doi.org/10.1002/ajmg.1320270417 [ Links ]
8. Beighton P, Wallis G, Viljoen D, Versfeld G. Osteogenesis imperfecta in southern Africa: Diagnostic categorisation and biomolecular findings. Ann N Y Acad Sci 1988;543(1):40-46. http://dx.doi.org/10.1111/j.1749-6632.1988.tb55314.x [ Links ]
9. Valadares ER, Carneiro TB, Santos PM, Oliveira AC, Zabel B. What is new in genetics and osteogenesis imperfecta classification? J Pediatr (Rio J) 2014;90(6):536-541. http://dx.doi.org/10.1016/j.jped.2014.05.003 [ Links ]
10. Alanay Y, Avaygan H, Camacho N, et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet 2010;86(4):551-559. http://dx.doi.org/10.1016/j.ajhg.2010.02.022 [ Links ]
11. Steinlein OK, Aichinger E, Trucks H, Sander T. Mutations in FKBP10 can cause a severe form of isolated osteogenesis imperfecta. BMC Med Genet 2011;12(1):152-155. http://dx.doi.org/10.1186/1471-2350-12-152 [ Links ]
12. Shaheen R, Al-Owain M, Faqeih E, et al. Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am J Med Genet A 2011;155A(6):1448-1452. http://dx.doi.org/10.1002/ajmg.a.34025 [ Links ]
13. Kelley BP, Malfait F, Bonafe L, et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res 2011;26(3):666-672. http://dx.doi.org/10.1002/jbmr.250 [ Links ]
14. Setijowati ED, van Dijk FS, Cobben JM, et al. A novel homozygous 5 bp deletion in FKBP10 causes clinically Bruck syndrome in an Indonesian patient. Eur J Med Genet 2012;55(1):17-21. http://dx.doi.org/10.1016/j.ejmg.2011.10.002 [ Links ]
15. Puig-Hervas MT, Temtamy S, Aglan M, et al. Mutations in PLOD2 cause autosomal-recessive connective tissue disorders within the Bruck syndrome-osteogenesis imperfecta phenotypic spectrum. Hum Mutat 2012;33(10):1444-1449. http://dx.doi.org/10.1002/humu.22133 [ Links ]
16. Venturi G, Monti E, Dalle Carbonare L, et al. A novel splicing mutation in FKBP10 causing osteogenesis imperfecta with a possible mineralization defect. Bone 2012;50(1):343-349. http://dx.doi.org/10.1016/j.bone.2011.10.023 [ Links ]
17. Barnes AM, Duncan G, Weis M, et al. Kuskokwim syndrome, a recessive congenital contracture disorder, extends the phenotype of FKBP10 mutations. Hum Mutat 2013;34(9):1279-1288. http://dx.doi.org/10.1002/humu.22362 [ Links ]
18. Caparros-Martin JA, Valencia M, Pulido V, et al. Clinical and molecular analysis in families with autosomal recessive osteogenesis imperfecta identifies mutations in five genes and suggests genotype- phenotype correlations. Am J Med Genet A 2013;161A(6):1354-1369. http://dx.doi.org/10.1002/ajmg.a.35938 [ Links ]
19. Schwarze U, Cundy T, Pyott SM, et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum Mol Genet 2013;22(1):1-17. http://dx.doi.org/10.1093/hmg/dds371 [ Links ]
20. Zhou P, Liu Y, Lv F, et al. Novel mutations in FKBP10 and PLOD2 cause rare Bruck syndrome in Chinese patients. PLoS One 2014;9(9):e107594. http://dx.doi.org/10.1371/journal.pone.0107594 [ Links ]
21. Umair M, Hassan A, Jan A, et al. Homozygous sequence variants in the FKBP10 gene underlie osteogenesis imperfecta in consanguineous families. J Hum Genet 2016;61(3):207-213. http://dx.doi.org/10.1038/jhg.2015.129 [ Links ]
22. Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 2001;68(4):978-989. http://dx.doi.org/10.1086/319501 [ Links ]
23. Henderson BD, Isaac N, Mabele O, Khiba S, Nkayi A, Mokoena T. Pamidronate treatment for osteogenesis imperfecta in black South Africans. S Afr Med J 2016;106(6 Suppl 1):S47-S49. http://dx.doi.org/10.7196/SAMJ.2016.v106i6.10992 [ Links ]
24. Wallis GA, Sykes B, Byers PH, Mathew CG, Viljoen D, Beighton P. Osteogenesis imperfecta type III: Mutations in the type I collagen structural genes, COL1A1 and COL1A2, are not necessarily responsible. J Med Genet 1993;30(6):492-496. http://dx.doi.org/10.1136/jmg.30.6.492 [ Links ]
25. Morello R, Bertin TK, Chen Y, et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006;127(2):291-304. http://dx.doi.org/10.1016/j.cell.2006.08.039 [ Links ]
26. Van Dijk FS, Nesbitt IM, Zwikstra EH, et al. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet 2009;85(4):521-527. http://dx.doi.org/10.1016/j.ajhg.2009.09.001 [ Links ]
27. Marini JC, Reich A, Smith SM. osteogenesis imperfecta due to mutations in non-collagenous genes: Lessons in the biology of bone formation. Curr Opin Pediatr 2015;26(4):50050-7. http://dx.doi.org/10.1097/MOP.0000000000000117 [ Links ]
28. Christiansen HE, Schwarze U, Pyott SM, et al. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet 2010;86(3):389-398. http://dx.doi.org/10.1016/j.ajhg.2010.01.034 [ Links ]
29. Viljoen D, Versfeld G, Beighton P. Osteogenesis imperfecta with congenital joint contractures (Bruck syndrome). Clin Genet 1989;36(2):122-126. http://dx.doi.org/10.1111/j.1399-0004.1989.tb03174.x [ Links ]
30. Mokete L, Robertson A, Viljoen D, Beighton P. Bruck syndrome: Congenital joint contractures with bone fragility. J Orthop Sci 2005;10(6):641-646. http://dx.doi.org/10.1007/s00776-005-0958-9 [ Links ]
 Correspondence:
Correspondence:
A Vorster
vorster@sun.ac.za
Accepted 6 February 2017


{kind=link}
{kind=link}