










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.107 n.5 Pretoria May. 2017
http://dx.doi.org/10.7196/samj.2017.v107i5.12447
CME
Diagnosis, monitoring and treatment of tuberous sclerosis complex: A South African consensus response to international guidelines
P J de VriesI; L LeclezioII; J M WilmshurstIII; G FieggenIV; E GottlichV, VI, VII; L JacklinVIII; I P NaikerIX; R NewajX; D ShamleyXI; B SchlegelXII, XIII; A VenterXIV
IMB ChB, MRCPsych, PhD Division of Child and Adolescent Psychiatry, Faculty of Health Sciences, University of Cape Town, South Africa
IIMSc (Med) Neurosci Division of Child and Adolescent Psychiatry, Faculty of Health Sciences, University of Cape Town, South Africa
IIIMB BS, MRCP, FC Paed (SA), MD; Tuberous Sclerosis Complex Clinic, Department of Paediatric Neurology, Red Cross War Memorial Children's Hospital and Faculty of Health Sciences, University of Cape Town, South Africa
IVMB ChB, MSc (Neurosci), FCS (SA) Neurosurgery, MD Division of Neurosurgery, Faculty of Health Sciences, University of Cape Town, South Africa
VMB BCh, DCH (SA), FC Paed (SA), Cert Nephrology (SA) Paed; Paediatric nephrologist, private practice, Johannesburg, South Africa
VIMB BCh, DCH (SA), FC Paed (SA), Cert Nephrology (SA) Paed; Department of Paediatric Nephrology, Faculty of Health Sciences, University of Pretoria, South Africa
VIIMB BCh, DCH (SA), FC Paed (SA), Cert Nephrology (SA) Paed; Clinical specialist, Discovery Health, South Africa
VIIIMB BCh, FCP (SA), MMed (Paed); Department of Paediatrics, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IXMB ChB, FRCP (Lond), FCP (SA); Nephrologist, private practice, Durban, South Africa
XMB BCh, FC Derm (SA); Dermatologist, private practice, Johannesburg, South Africa
XIMB BCh, FCP (SA); Neurologist, private practice, Johannesburg, South Africa
XIIMB ChB, FC Paed (SA) Tuberous Sclerosis Complex Clinic, Department of Paediatric Neurology, Red Cross War Memorial Children's Hospital and Faculty of Health Sciences, University of Cape Town, South Africa
XIIIMB ChB, FC Paed (SA) Paediatric neurologist, private practice, Cape Town, South Africa
XIVMB ChB, MMed, PhD, DCH (SA), FC Paed (SA) Department of Paediatrics and Child Health, Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
ABSTRACT
Tuberous sclerosis complex (TSC) is a genetic disorder with multi-system manifestations and a high burden of disease. In 2013, an international panel of TSC experts revised the guidelines for the diagnosis, surveillance and treatment of the disorder. In South Africa (SA), a local multidisciplinary group of healthcare professionals and TSC researchers reviewed the international guidelines to generate an SA consensus clinical update on the identification, diagnosis, treatment and lifelong monitoring of individuals who live with TSC. We strongly endorse dissemination and use of the international guidelines for the assessment, monitoring and treatment of TSC. In addition, we strongly support access to genetic testing and to mTOR (mammalian target of rapamycin) inhibitors to treat subependymal giant cell astrocytomas not amenable to surgery and renal angiomyolipomas larger than 3 cm, and as adjunctive treatment for refractory focal seizures. We await with interest results from mTOR inhibitor trials of skin and TSC-associated neuropsychiatric disorders (TAND). With regard to training, we recommend the inclusion of TSC in undergraduate and postgraduate medical and health sciences curricula, and the promotion of other continuing professional development events to raise awareness about TSC. We also support the development of a TSC user/carer/parent organisation to provide an informal support network for families across SA. We acknowledge that some progress has been made in recent years in SA, but much remains to be done. We hope that this SA consensus clinical update based on the international guidelines will make a positive contribution to increase knowledge and improve clinical care for all patients who live with TSC in SA, and their families.
Tuberous sclerosis complex (TSC) is a multi-system genetic disorder with a birth incidence around 1:5 800.[1,2] It affects ~1.5 million people around the globe with no obvious differences in prevalence based on gender or ethnicity, although epidemiological data from Africa, including South African (SA) populations, are lacking.[1,2] TSC can affect almost any organ system in the body, including the brain, heart, skin, kidneys and lungs. The greatest morbidity and mortality is associated with neurological, neuropsychiatric and renal manifestations.[2] To reduce the morbidity and mortality in TSC, evidence-based management and co-ordination of care across medical specialties is crucial throughout the lifespan of the patient.[2,3]
TSC was first described in 1880 by the French neurologist Bourneville at the Salpetriere Hospital in Paris. Bourneville had a 15-year-old patient with severe intellectual disability and intractable epilepsy. After the death of the child, postmortem examination revealed unusual white, hard lesions on the cortical surface of the brain, and multiple small nodules lining the ventricular surface of the brain. Bourneville described these white, hard lesions as 'sclereuse tubereuse (white, potato-like lesions) of the cerebral cor-tex.[2] Previously used terms for TSC include 'Bourneville's disease' and 'epiloia but use of these terms is now strongly discouraged.
The early diagnostic criteria, first documented in 1979, were based purely on the systematic clinical observations of Dr Manuel R Gomez, a physician at the Mayo Clinic in Rochester, USA. [4,5] Gomez identified that patients with TSC had a wide range of physical manifestations across almost all organ systems, including the brain, skin, heart, eyes, kidneys and lungs, and that multi-system involvement was highly variable between patients. He also recognised (in contrast to the late 19th-century descriptions, where all patients were believed to have intellectual disability) that the intellectual abilities of individuals with TSC were extremely varied, ranging from normal intellectual ability to profound intellectual disability. Gomez's observations remain at the heart of the diagnostic criteria and treatment guidelines for TSC, even in the most recent international revisions.[3,6]
Very little was understood about the pathophysiology of TSC until the 1990s and 2000s. The TSC1 gene (on chromosome 9q34) and the TSC2 gene (on chromosome 16p13.3) were identified in 1993 and 1997, respectively, but it remained unclear how their protein products functioned intracellularly.[1,2] We now know that the TSC1 and TSC2 proteins form an intracellular complex in all cells, upstream to a protein called the mammalian target of rapamycin, typically referred to by its abbreviation mTOR. Two of the key intracellular functions of mTOR are protein synthesis and regulation of cell migration and proliferation. In a situation where either the TSC1 or TSC2 gene is affected due to a mutation, the TSC1-TSC2 complex is therefore not able to inhibit or 'put the brakes' on the mTOR complex, leading to overgrowth in specific cells, causing the benign tumours that are seen in individuals with TSC.[2,7,8] Recognition of this fundamental role of the TSC proteins in cells has also led to the discovery of molecularly targeted treatments for TSC in the form of mTOR inhibitors. These agents reduce the 'overactivation' of mTOR that results from disruption of the TSC1-TSC2 complex.[2,7,8] Fig. 1 shows a graphic representation of intracellular signalling in TSC, indicating where the mTOR inhibitors have their molecular treatment target.

In about 70% of cases, TSC occurs due to 'sporadic' mutations (de novo). In the remaining 30% of cases, the mutation is inherited in an autosomal dominant fashion, and is therefore 'familial'. Individuals with TSC have a 50% chance of passing the mutated TSC gene on to their children. Unaffected parents with one child with TSC have an approximately 1% chance of having another child with TSC.[1-3]
The clinical presentation and needs of individuals with TSC
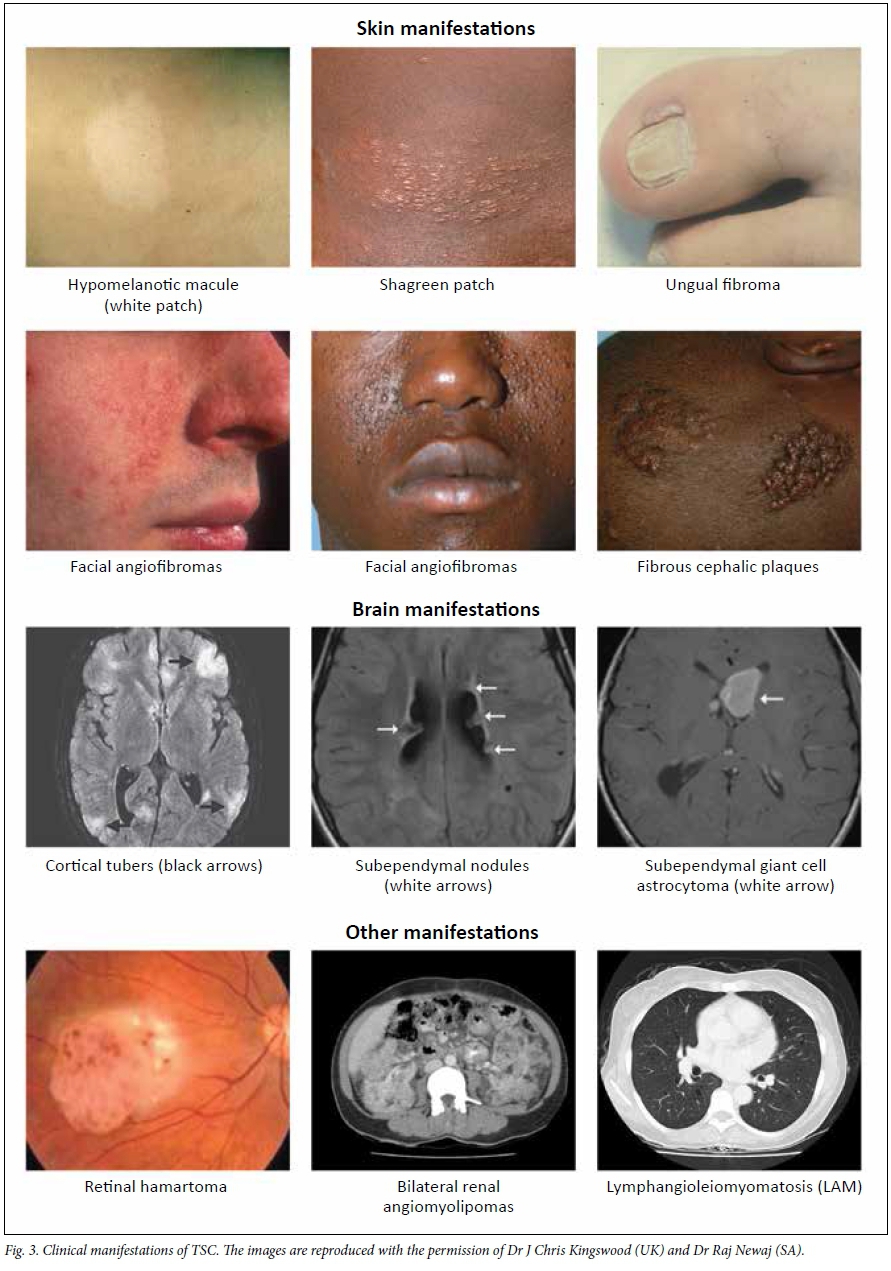
Patients with TSC can have a complex and changeable clinical presentation. Firstly, many different organ systems can be involved, but in a variable way, termed variable expression. For instance, some patients may have significant skin involvement but few brain manifestations; others may have little skin involvement but significant renal and brain involvement.
Secondly, TSC manifestations may present at different ages, termed age-related expression. For instance, cardiac rhabdomyomas may be identified with antenatal fetal ultrasound or in the first year of life. After that they tend to regress and disappear. In contrast, skin and renal manifestations are typically not present at birth, but gradually appear over the first 5 - 15 years of life.[1,2]
Given the variability of manifestations and the age-related expression, individuals with TSC may present to a range of healthcare professionals with signs and symptoms of possible TSC. Table 1 shows a list of clinical manifestations that may suggest possible TSC in a new patient. In the antenatal period, a fetus is most likely to be identified with possible TSC when cardiac rhabdomyomas are identified on ultrasound. There are also cases where central nervous system manifestations (such as cortical tubers) are identified antenatally. In the first few years of life, the most typical presentation of TSC is likely to be with seizures, in particular focal seizures and infantile (epileptic) spasms, often with an onset in the first 12 months of life. Many parents notice unusual jerky movements in their 2 - 9-month-old infant, and these spasm-like movements are often confused with colic. Clear-cut seizures typically lead to hospitalisation, neurological work-up and clinical diagnosis of TSC in the presence of at least two major diagnostic criteria of TSC (see later in the article).

After the first few years of life, children may present with skin manifestations such as facial angiofibromas or hypomelanotic macules (white patches), or with developmental or behavioural concerns. Autism spectrum disorder is seen in 40 - 50% of individuals with TSC, and TSC is seen in as many as 4% of children with autism spectrum disorder. For this reason, all children with autism spectrum disorder and epilepsy should be evaluated for possible TSC. Individuals with mild physical manifestations of TSC may present with skin, academic or mental health problems, or be identified when renal manifestations are picked up on an ultrasound/magnetic resonance imaging (MRI) scan by chance.
Individuals with TSC may develop a benign brain tumour called a subependymal giant cell astrocytoma (SEGA) that almost invariably arises in the lateral ventricle adjacent to the foramen of Monro and may present with raised intracranial pressure due to obstructive hydrocephalus. It is therefore possible that presentation and diagnosis of a SEGA will lead to comprehensive clinical work-up and diagnosis of TSC.
Adolescent boys or young men with TSC are sometimes diagnosed when they have a skin rash (often misdiagnosed as acne) that causes profuse bleeding during shaving. Some young people and adults present with ungual growths on their hands or feet that cause embarrassment or discomfort.
A proportion of mildly affected individuals with TSC are only diagnosed when they have a child diagnosed with TSC, or when there is a family history of TSC and a child receives comprehensive physical work-up.[1-3,6,8]
Given the variability of manifestations and the age-related onset, it is clear that individuals with TSC have both clinically complex and multi-system needs that will require input from a range of healthcare professionals across their lifespan. Patients therefore require a multidisciplinary diagnostic, monitoring and treatment approach using evidence-based intervention strate-gies.[2,3,8] In SA, the majority of patients with TSC are likely to present to and be managed by general practitioners rather than by specialists. For this reason, it is of utmost importance for generalist and primary care teams to be aware of the best-practice guidelines for the assessment, monitoring and treatment of TSC.
Revision of the international diagnostic criteria and consensus guidelines for monitoring and treatment of TSC
As listed above, the first set of diagnostic criteria were generated by Gomez in 1979, based on his observations at the Mayo Clinic. By 1998, the TSC clinical and research community had grown, and a decision was made to have a conference in the USA with the aim of revising and generating international consensus on diagnostic criteria and treatment. The meeting was attended by 23 TSC experts from four countries, and recommendations were based on majority views of expert opinion. The meeting resulted in publication of the first set of consensus diagnostic criteria,[4] often referred to as the 'Roach criteria'. In addition, a consensus document on treatment and monitoring was published.[5]
The genes for TSC had been identified by the time of the 1998 consensus conference, but little more was known about the biology of TSC, and no disorder-specific treatments were available. In 2003, the link between TSC1-TSC2 and mTOR was established, which rapidly led to clinical trials of mTOR inhibitors for TSC. In 2006, David Franz and colleagues in Cincinnati showed that rapamycin (the prototypical mTOR inhibitor) could shrink SEGAs in TSC.[2] By 2012, everolimus, another mTOR inhibitor, had received Federal Drug Administration and European Medicines Agency approval for the treatment of brain (SEGAs) and renal (angiomyolipoma) lesions in TSC.[2]
With the support of the National Institutes of Health (a research funding agency of the US Department of Health) and the Tuberous Sclerosis Alliance (a US advocacy and support organisation), a second consensus conference was convened in 2012, attended by 79 TSC clinicians, researchers, individuals with TSC and parents, from 14 countries. The majority of the recommendations were based on evidence from peer-reviewed publications. This meeting resulted in a revised set of diagnostic criteria[6] and an enhanced set of management and surveillance guidelines.[3] Full details of the processes and procedures to generate relevant literature, review and weigh up (grade) evidence were presented clearly in the two core publications.[3,6] To summarise in brief, 12 subcommittees were organised to focus on specific disease areas. Subcommittees included a range of clinicians, researchers and parents/individuals with TSC. Each subcommittee had about 12 months to identify relevant questions, review the relevant literature, evaluate and weigh up the strength of data, and present an initial set of recommendations. Where consensus recommendations could not be made based on conflicting or lack of evidence, subcommittees were asked to provide recommendations for future research to answer these unresolved questions. A centralised literature search was performed for all subcommittees to use. The search included PubMed and Scopus databases between 1997 and 2012 (the year of the review). A total list of 2 692 articles were retrieved and provided to subcommittees. Subcommittees were also encouraged to add additional key literature based on specific subcommittee questions, if data provided in the central literature were insufficient. Evidence was evaluated and weighed up (graded) using the evidence-based framework of the National Comprehensive Cancer Network Clinical Guidelines (all details provided in references).[3,6] Category 1 recommendations were based on high-level evidence and uniform consensus, category 2 recommendations were based on lower-level evidence and consensus, and category 3 recommendations were made where consensus could not be reached, regardless of evidence. All subgroups presented their recommendations at a 2-day overall consensus workshop where final consensus recommendations were made, from which the international guidelines were drawn up. All final recommendations were graded in the international publications. Interested readers are encouraged to read these documents.[3,6]
The diagnostic criteria and management guidelines were adopted internationally by TSC parent/user organisations and clinical groups, but the international consensus panel encouraged regional and national groups to consider the application and implementation of guidelines in local settings.
TSC in South Africa
Based on birth incidence and prevalence estimates for TSC and a midyear population estimate of 54 million South Africans in 2014, there are likely to be somewhere between 5 000 and 10 000 people with TSC in SA. While these numbers may not sound significant in the context of our many other health challenges, the complex, lifelong and multisystem nature of TSC leads to an extremely high burden of disease, a high burden of care and treatment, and a very significant impact on disability and health economics.
However, very little is known about TSC in SA. There are no epidemiological data on the prevalence or incidence of TSC in this country, and no national registry or national organisation exists. Red Cross War Memorial Children's Hospital in Cape Town has, to our knowledge, the only dedicated TSC clinic in the country, which includes ~80 - 100 patients. Collectively, the TSC experts and consensus working group members who drafted this consensus clinical update are involved in the monitoring and treatment of no more than 200 patients across SA. This observation suggests that the majority of individuals with TSC in SA are cared for in either general practice or non-specialist settings in the state or private sector, or may not even have reached medical services to receive diagnosis and treatment.
It is unclear how much healthcare professionals in SA know about TSC. Given the fact that it is a rare disease, many healthcare professionals may not be exposed to patients with TSC during their undergraduate or clinical training, or even during specialisation. Many nurses, psychologists, occupational and speech and language therapists, and other community-based practitioners are unlikely to receive training on recognition of the clinical manifestations of TSC. There is a great need for multidisciplinary teams in SA for the assessment, monitoring and treatment of the disorder.
Two of the key changes in the 2013 diagnostic and treatment guidelines for TSC involved the introduction of molecular genetic testing as a 'genetic diagnostic criterion' for TSC, and introduction of mTOR inhibitors for the treatment of specific manifestations of the disorder.[3,6] There are at present no clinical laboratories in SA that can do mutation analysis to establish a molecular diagnosis of TSC. Samples have to be sent to the USA, the UK or Europe for clinical molecular genetic testing. No mTOR inhibitors have received marketing authorisation in SA for any TSC-related indications to date. We are, however, aware of a number of individuals with TSC who are currently receiving mTOR inhibitors 'off-label' for various specific TSC indications.
The primary purpose of this SA consensus response to the international guidelines was to promote awareness of TSC among colleagues at primary healthcare level and specialists who may be involved in the care of affected individuals. Given that a well-considered set of international guidelines had already been drawn up and published, we elected not to generate our own guidelines but rather to prepare a consensus clinical update in response to the internationally adopted guidelines.[3,6]
SA consensus clinical update in response to the 2013 revised diagnostic criteria and international guidelines for TSC
Given the clinical need of patients with TSC and the relative lack of expertise on TSC in SA, a decision was made to convene an SA consensus panel with the aim of developing a formal response to the international guidelines. The panel was led by PJdV, an international TSC expert, and included healthcare professionals with experience in the management of TSC from across SA. In the context of the two-tier healthcare system in the country, we deliberately included clinicians from both the state and private sectors. All panel members were provided with the two consensus articles[3,6] and were asked to review all recommendations. Each panel member was asked to indicate agreement or disagreement with each of the recommendations independent of other experts, and the results were collated by one of the authors (LL). A face-to-face consensus meeting took place on 17 July 2014 in Johannesburg, with all the authors except one (BS) present. At the consensus meeting, the background and rationale of the meeting was presented by PJdV, all areas of consensus and disagreement were discussed, and a plan of action for next steps was generated.
Overall, the SA consensus group endorsed the two articles, with comments and caveats about resource implications and implementation into practice as outlined below.
A clinical flowchart for the diagnosis, monitoring and treatment of TSC (Fig. 2)

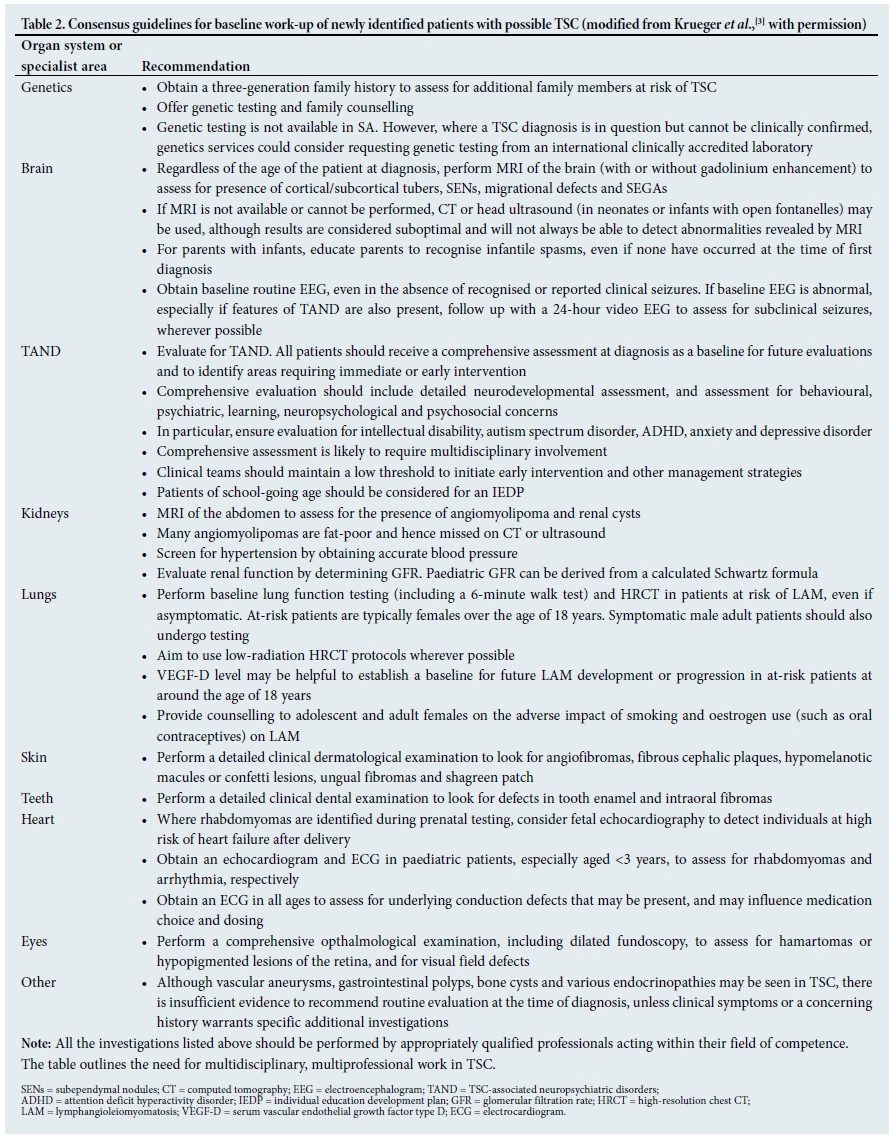
Once the first clinical suspicion of TSC has presented (see Table 1 for examples), patients require a comprehensive physical work-up as outlined in Table 2.[3] The primary purpose of clinical assessment is to determine whether a patient with 'possible' TSC has sufficient clinical manifestations to meet criteria for 'definite' TSC. The diagnostic criteria are presented in Table 3.[6] As highlighted above, a molecular diagnosis of TSC can now be made when an accredited clinical laboratory identifies a disease-associated mutation in TSC1 or TSC2. Many mutations identified in clinical molecular laboratories around the world are of unclear significance, and may represent non-disease-associated polymorphisms (i.e. changes in the DNA sequence with an unclear consequence with regard to protein structure and/or function).
Given that SA has no clinical laboratories that can do mutation analysis for TSC, the consensus panel recommended that efforts should be made to introduce molecular genetic testing into a suitable SA clinical laboratory. In the meantime, SA clinicians should use the clinical diagnostic criteria to determine whether a patient meets the criteria for the disorder.
A patient meets the criteria when they have clinical evidence of two major diagnostic criteria for TSC (Table 3) or if they meet one major and two or more minor criteria. Fig. 3 illustrates some of the common diagnostic characteristics of TSC.
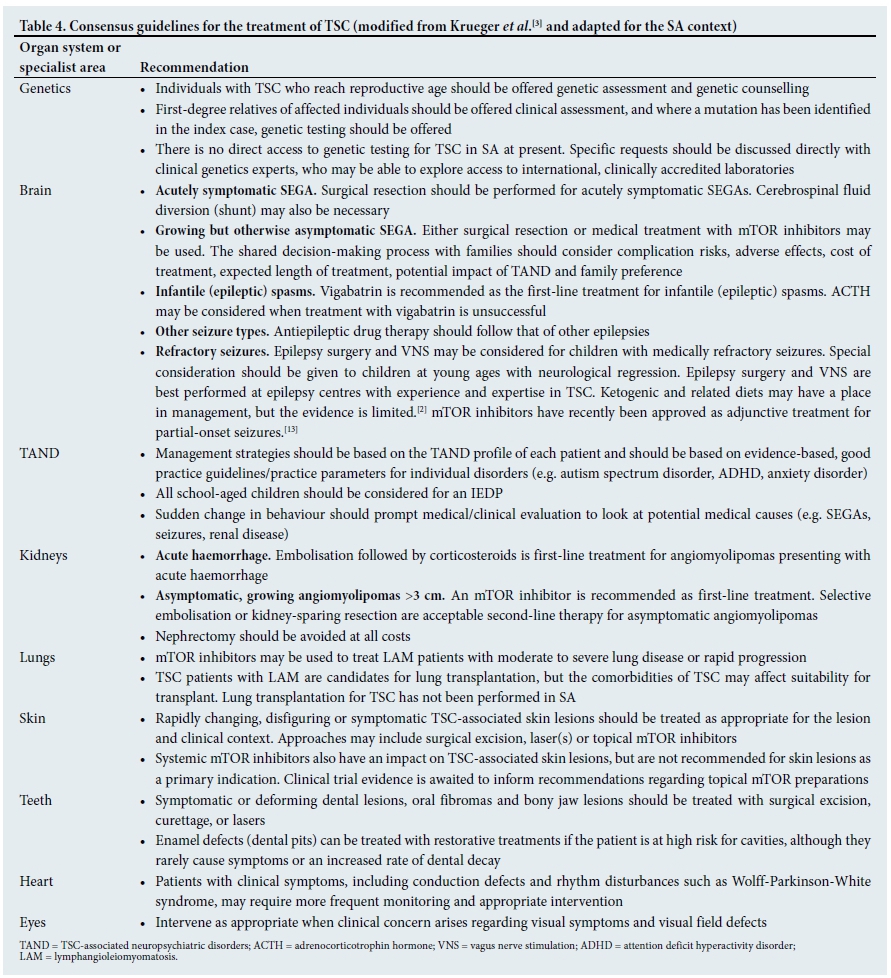
Once a patient has been diagnosed with TSC, they will require appropriate treatment based on their clinical manifestations and needs. Table 4 sets out the international treatment guidelines for TSC.[3]
The SA expert panel recognised that a number of the treatment and monitoring recommendations may be hard to access in a resource-limited SA setting, such as in accessing annual MRI scans of the brain, or electro-encephalograms. However, we were encouraged to hear that some SA centres were able to follow the international consensus recommendations. We therefore agreed that these international guidelines should be seen as an aspirational set of recommendations.
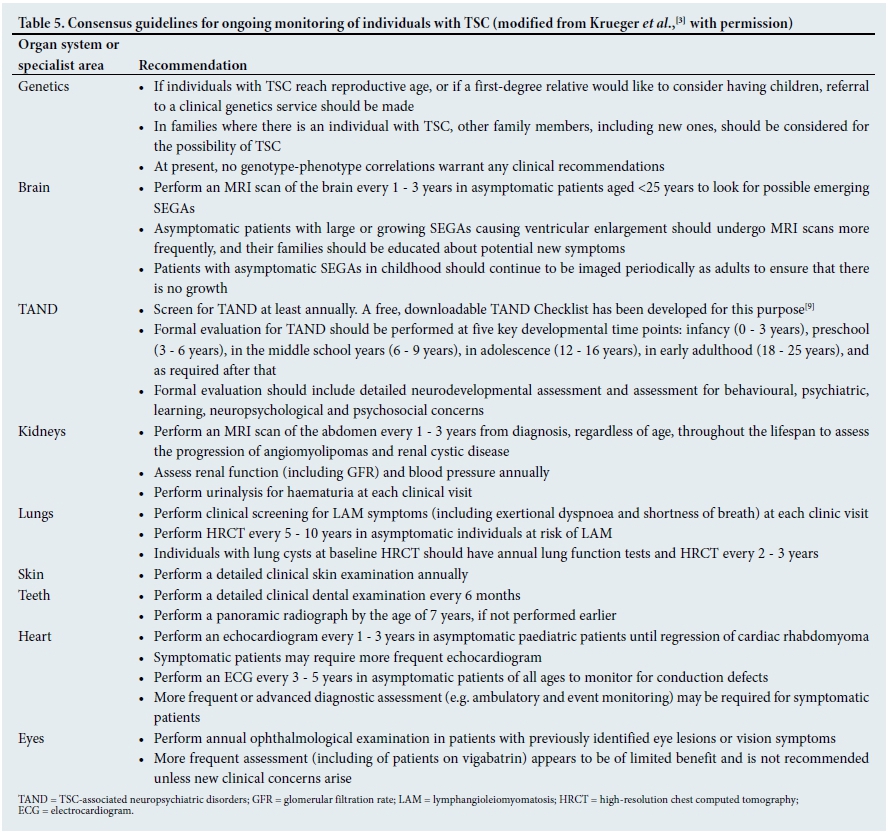
Ongoing monitoring of all patients with TSC is required, even if they had no overt problems or concerning manifestations at a previous clinic visit. Given the known age-related expression, patients may develop a range of clinical difficulties over a relatively short period of time. The international guidelines for regular monitoring of patients with TSC are presented in Table 5.[3]
Rapid assessment and treatment is required where patients present with sudden changes in clinical presentation. Examples of 'change' may include changes in seizure control or pattern, appearance of focal neurological signs, deterioration in school performance, or behavioural change (e.g. an individual who has a change in sleep or mood or becomes disruptive without obvious reason). Physical work-up should be prioritised and careful consideration given to the possibility of a SEGA, renal complications or other physical or metabolic abnormality.
Frequently asked clinical questions about diagnosis, monitoring and treatment in SA
During the preparation of this article, a number of questions emerged from within the consensus panel and from other clinical stakeholders in SA. We address some of the key questions here.
• Do we need to do MRI scans of the abdomen every 1 - 3 years, or is there a safe alternative in SA? Given that the majority of patients with TSC will develop renal angiomyolipomas associated with hypertension, pain and risk of haemorrhage, early identification and treatment of renal lesions is essential. Identification of renal lesions is therefore the primary reason for abdominal imaging. In order to understand and treat the renal lesion, we need to know what is there and how big it is. TSC renal lesions are often of odd shapes and fat-poor, making it very difficult to visualise and measure their size accurately. Ultrasound has been assessed in the literature, and has been shown not to be sufficiently accurate for use in TSC and polycystic disease. Apart from renal lesions, it is also important to detect additional TSC-related findings in the liver, aorta/blood vessels, spleen, pancreas, etc., all of which can be identified in patients with TSC during MRI surveillance of the kidneys. These TSC manifestations are not detected or evaluated with renal ultrasound. For these reasons, the evidence-based recommendation for renal and abdominal monitoring is abdominal MRI every 1 - 3 years.
• Do we need an MRI scan of the brain every 1- 3 years, or will a computed tomography (CT) scan be sufficient? CT scans are not recommended in TSC for two reasons: (i) the need for serial imaging from early childhood into adulthood poses real concern about unnecessary cumulative exposure to ionising radiation when a non-exposure alternative (MRI) exists; and (ii) MRI is superior to CT at detecting soft-tissue components of SEGAs, especially when tumours are only partially calcified. While a SEGA may be diagnosed on a CT scan, especially if there is a calcified component, MRI with post-contrast sequences is better able to define the tumour margin and the relationship to important structures such as the foramen of Monro, fornix and blood vessels, and is also a more reproducible means of measuring and monitoring tumour size over time.
• Can we combine MRI of the abdomen and brain in a single session? We strongly recommend that MRI of the abdomen and brain is co-ordinated between neurology, nephrology and radiology teams to ensure that patients only need to come for a single MRI session. This may be particularly use ful for patients who require sedation or an anaesthetic for the procedure.
• How important are neurodevelopmental assessments in TSC? Many people with TSC have neurodevelopmental difficulties, but many do not receive regular and detailed assessments. For this reason, we strongly support the international recommendations to perform annual screening for neurodevelopmental difficulties as part of a broader evaluation of all TSC-associated neuropsychiatric disorders (TAND).[3,9] We also strongly support the international recommendation to perform regular detailed neurodevelopmental and mental health assessments at key timepoints, as shown in Table 5.
• Is it important to screen for and assess for autism spectrum disorder? TSC is the medical condition most strongly associated with autism spectrum disorder. Almost 50% of people with TSC meet criteria for autism spectrum disorder, and many have social-communication difficulties even if they do not meet criteria for autism spectrum disorder. For this reason, it is very important to screen for autism spectrum disorder from early on, and to do a comprehensive diagnostic work-up if there are any concerns. The TAND Checklist[9,10] was designed as a simple pen-and-paper tool to help clinicians screen for autism spectrum disorder and other TAND.
• Are formal assessments required to assist with school placements for children with TSC? Even though children with TSC can have a huge range of intellectual abilities, the rates of scholastic difficulties are very high.[2,9] Many children with TSC, even when they have normal or above-average IQs, have specific learning disorders that will affect their ability to achieve in school. It is also important to remember that any aspect of TAND can affect a child's ability to access learning, e.g. attention deficit hyperactivity disorder (ADHD), autism spectrum disorder, anxiety or depressive disorders, specific neuropsychological deficits, and psychosocial factors. For this reason, we strongly support the international recommendations that all school-aged children should be assessed for their educational needs and for the potential need for an individual education development plan (IEDP) (Table 4).
Recommendations for next steps to support individuals with TSC in SA
Given the discrepancy between needs of patients with TSC and available resources and expertise in SA, the SA consensus panel made a number of recommendations for implementation in SA (Table 6). We strongly endorsed the dissemination and use of the international guidelines for the assessment, monitoring and treatment of TSC. In addition, we strongly support access to genetic testing, which is increasingly used around the world in clinical genetics settings to do preimplantation testing, and to examine the association between genotype and phenotype. At present, there are no clinical recommendations regarding differences between TSC1 and TSC2 mutations, but there is significant research interest in the use of genotypic information to predict phenotype and potential for treatment response.[11,12]
We also strongly advocate for the relevant regulatory bodies in SA to provide marketing authorisation and reimbursement to support access to mTOR inhibitors for patients in SA for all internationally approved indications. Everolimus, one of the mTOR inhibitors, received marketing authorisation in the USA and Europe for the treatment of SEGAs not amenable to surgery, and for renal angiomyolipomas larger than 3 cm.[3] Recently, everolimus also received marketing authorisation in Europe for the adjunctive treatment of refractory focal seizures, following results of an international multi-centre trial.[13] In addition, topical preparations are used for facial angiofibromas and other skin manifestations with very positive outcomes. There is also evidence that systemic mTOR inhibition improves skin manifestations in patients who take mTOR inhibitors for SEGAs or angiomyolipomas. A number of clinical trials are underway exploring the potential for mTOR inhibitors to improve TAND such as autism, intellectual disability, academic difficulties or specific neuropsychological deficits.[2]
Apart from clinical aspects of TSC, we also recommend its inclusion in undergraduate and postgraduate medical and health sciences curricula, and the promotion of other continuous professional development events to raise awareness about it. We support the development of a TSC user/carer/parent organisation that can develop into an informal support network for families across SA. TSC International (TSCi) is a worldwide network of user/carer organisations (http://www.tscinternational.org), and it would be a very powerful strategy to connect the SA TSC community to TSCi. We strongly support ongoing research on TSC in SA and other African countries, given the clear need, but lack of local research data.
In high-income settings, it is standard practice for an individual with TSC to be seen by a range of specialists, including paediatric neurology, dermatology, paediatric nephrology, psychiatry, psychology and so on. In an SA setting, we acknowledge that, owing to resource limitations, many patients with TSC may currently be cared for in primary or secondary care settings. We would, however, like to encourage referral to and appropriate liaison with tertiary services, particularly to teams with a particular interest in and experience working with individuals who have TSC. Patients with SEGAs, angiomyolipomas, hypertension, dermatological manifestations, epilepsy and neuropsychiatric disorders should always be referred to specialists and, wherever possible, to tertiary settings. While general practitioners in SA may have a central role in the overall co-ordination of care, we do not recommend that a general practitioner be expected to carry out clinical management of an individual with TSC in isolation. In SA, a shared-care approach with clear communication and liaison between generalists and specialists to build a virtual 'team around the family', with families as active and equal partners in this process, would be ideal.
We acknowledge that some positive progress has been made in recent years. A number of talks on TSC have been presented at local and national fora, and a number of SA-based publications have been produced.[10,14,15] In addition, SA is a participating country in the International Natural History Study of TSC, referred to as TOSCA (TuberOus SClerosis Registry to increase disease Awareness).[16] We sincerely hope that these SA guidelines will also make a positive contribution to increase knowledge and improve clinical care for all patients who live with TSC in SA, and their families.
Acknowledgements. The SA consensus meeting was sponsored by Novartis. We are grateful to Novartis colleagues Dr Michelle Blou, Medical Director, and Dr Chris Nathaniel, Oncology Medical Advisor, for funding and logistic support for the meeting. The sponsors did not play any role in the content or conduct of the meeting, and had no influence on or input in the consensus clinical update manuscript prepared by the consensus working group. No science writers were used to produce the manuscript.
Author contributions. All authors contributed to the consensus meeting and to generation of the outline for the article. PJdV prepared the first draft of the article. All authors contributed to drafts of the article and to reviewer comments. All authors approved the final version of the article.
Funding. The manuscript was written without any financial support from Novartis or any other organisation.
Conflicts of interest. All the authors were members of the consensus meeting sponsored by Novartis and received honoraria and travel for participation. PJdV is a study steering group member of three clinical trials of mTOR inhibitors, sponsored by Novartis, and a co-principal investigator on two investigator-initiated phase II clinical trials of mTOR inhibitors, part-funded by Novartis, Autism Speaks, the TS Alliance, Tuberous Sclerosis Association and the Wales Gene Park. PJdV is also a member of the Working Committee and Scientific Advisory Board of the TOSCA natural history study, sponsored by Novartis. He was chairman of the Neuropsychiatry Panel at the 2012 International Consensus Conference and contributed to the two manuscripts produced by the meeting.
LL has a Career Development Studentship funded by the Tuberous Sclerosis Association (UK).
JMW is PI for the Red Cross War Memorial Children's Hospital (RCWMCH) site for the TOSCA study sponsored by Novartis. Proceeds from this study are used to support the RCWMCH TSC service. JMW is also chair of the Pediatric Commission for the International League Against Epilepsy, secretary of the International Child Neurology Association, education officer for the Commission on African Affairs and executive board member of the African Child Neurology Association and the Paediatric Neurology and Developmental Association of Southern Africa. EG is a part-time employee of Discovery Health, where he is responsible for the development and implementation of the KidneyCare Programme aimed at managing members of the scheme with chronic kidney disease. AV is an advisory board member for ADHD medications, funded by Novartis. GF, EG, LJ, IPN, RN, DS and BS did not declare any additional conflicts of interest.
References
1. Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet 2008;372(9639):657-668. http://dx.doi.org/10.1016/S0140-6736(08)61279-9 [ Links ]
2. Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatry aspects of tuberous sclerosis complex. Lancet Neurol 2015;14(7):733-745. http://dx.doi.org/10.1016/S1474-4422(15)00069-1 [ Links ]
3. Krueger DA, Northrup H, Roberds S, et al. Tuberous sclerosis complex surveillance and management: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49(4):255-265. http://dx.doi.org/10.1016/j.pediatrneurol.2013.08.002 [ Links ]
4. Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: Revised clinical diagnostic criteria. J Child Neurol 1998;13(12):624-628. http://dx.doi.org/10.1177/088307389801301206 [ Links ]
5. Roach ES, DiMario FJ, Kandt RS, et al. Tuberous Sclerosis Consensus Conference: Recommendations for diagnostic evaluation. J Child Neurol1999;14(6):401-407. http://dx.doi.org/10.1177/088307389901400610 [ Links ]
6. Northrup H, Krueger DA, Roberds S, et al. Tuberous sclerosis complex diagnostic criteria update: Recommendations from the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49(4):243-254. http://dx.doi.org/10.1016/j.pediatrneurol.2013.08.001 [ Links ]
7. De Vries PJ, Howe CJ. The tuberous sclerosis complex proteins - a GRIPP on cognition and neurodevelopment. Trends Mol Med 2007;13(8):319-326. http://dx.doi.org/10.1016/j.molmed.2007.06.003 [ Links ]
8. De Vries PJ. Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics 2010;7(3):275-282. http://dx.doi.org/10.1016/j.nurt.2010.05.001 [ Links ]
9. De Vries PJ, Whittemore VH, Leclezio L, et al. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol 2015;52(1):25-35. http://dx.doi.org/10.1016/j.pediatrneurol.2014.10.004 [ Links ]
10. Leclezio L, Jansen A, Whittemore VH, et al. Pilot validation of the tuberous sclerosis-associated neuropsychiatry disorders (TAND) checklist. Pediatr Neurol 2015;52(1):16-24. http://dx.doi.org/10.1016/j.pediatrneurol.2014.10.006 [ Links ]
11. Van Eeghen AM, Black ME, Pulsifer MB, et al. Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet 2012;20(5):510-515. http://dx.doi.org/10.1038/ejhg.2011.241 [ Links ]
12. Wong HT, McCartney DL, Lewis JC, et al. Intellectual ability in tuberous sclerosis complex correlates with predicted effects of mutations on TSC1 and TSC2 proteins. J Med Genet 2015;52(12):815-822. http://dx.doi.org/10.1136/jmedgenet-2015-103154 [ Links ]
13. French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): A phase 3, randomized, double-blind, placebo- controlled study. Lancet 2016;388(10056):2153-2163. http://dx.doi.org/10.1016/S0140-6736(16)31419-2 [ Links ]
14. Samia P, Donald KA, Schlegel B, et al. Parental understanding of tuberous sclerosis complex. J Child Neurol 2015;30(10):1281-1286. http://dx.doi.org/10.1177/0883073814558121 [ Links ]
15. Lachman A, van der Merwe C, de Vries P. Cognitive and functional outcomes after a trial of an mTOR inhibitor in an adolescent with neuropsychiatric sequelae of TSC. Eur Psychiatry 2016;33:S355. http://dx.doi.org/10.1016/j.eurpsy.2016.01.1265 [ Links ]
16. Kingswood JC, Bruzzi P, Curatolo P, et al. TOSCA - first international registry to address knowledge gaps in the natural history and management of tuberous sclerosis complex. Orphanet J Rare Dis 2014;9:182. http://dx.doi.org/10.1186/s13023-014-0182-9 [ Links ]
 Correspondence:
Correspondence:
P J de Vries
petrus.devries@uct.ac.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}