










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.107 no.3 Pretoria mar. 2017
http://dx.doi.org/10.7196/samj.2017.v107i3.11332
IN PRACTICE
CASE REPORT
A review of patients with glutaric aciduria type 1 at Inkosi Albert Luthuli Central Hospital, Durban, South Africa
R GovenderI; A MithaII; L MubaiwaI
IFCPaed (SA); Department of Paediatric Neurology, Faculty of Medicine, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
IIFCRad (SA); Department of Radiology, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
ABSTRACT
Glutaric aciduria type 1 (GA1) is an organic acidaemia. The objective of this study was to describe the profile of patients diagnosed with GA1 at Inkosi Albert Luthuli Central Hospital, Durban, South Africa from 2007 to 2015. We identified 6 children (4 girls, 2 boys) in a retrospective review. The mean age at diagnosis was 12 months. Clinical findings on presentation were encephalopathic crises (n=4), hypotonia (n=4) and macrocephaly (n=5). Other complications included seizures (n=4), dystonia (n=3) and bulbar dysfunction (n=4). Urine organic acid screens showed elevated glutaric acid levels (n=6). Five patients tested positive for the A293T mutation on the glutaryl-co-enzyme A (CoA) dehydrogenase gene. Abnormalities on magnetic resonance imaging screening included hyperintense basal ganglia ( n=6), widened perisylvian fissures (n=6), and an abnormal signal in the cerebral peduncles (n=5) and central tegmental tract (n=4). All patients were treated with L-carnitine and dietary modification. Two patients had a static clinical course, 1 patient gained milestones, and 3 have shown further neuroregression.
Glutaric aciduria type 1 (GA1) is a rare, autosomal-recessive organic acidaemia. It is caused by deficiency of glutaryl-co-enzyme A (CoA) dehydrogenase (GCDH) resulting from a mutation in the GCDH gene on chromosome 19p13.2. There are 108 known disease-causing mutations in the Human Gene Mutation Database.[1] The GCDH enzyme is vital in the final degradation of lysine, L-hydroxylysine and L-tryptophan. A deficiency of GCDH leads to accumulation of glutaric acid (GA) and 3-hydroxyglutarate (3-OHGA) in the brain. Neurotoxicity affecting the basal ganglia is caused by mitochondrial damage from α-ketoglutarate depletion provoked by excess GA.
The overall global prevalence of GA1 is 1/100 000.[2,3] However, the prevalence of the A293T mutation in South African (SA) patients of black indigenous ancestry is >1/100 000 and has been documented as 1/5 184.[4]
The typical clinical picture of GA1 is that of a developmentally normal infant with macrocephaly, who presents with an encephalo-pathic crisis following an intercurrent illness. The clinical course is characterised by neuroregression, dystonia and seizures. Treatment comprises a low-lysine diet, carnitine supplementation, and aggressive management of intercurrent illnesses and metabolic crises. Early diagnosis is critical, as aggressive management in the presymptomatic period prevents progressive neurological damage.
Methods
The main objective of the study was to describe the profile of patients with GA1. Furthermore, we sought to raise awareness of the condition among health professionals in SA. All patients diagnosed with GA1 at Inkosi Albert Luthuli Central Hospital, Durban, SA between 2007 and 2015 were included in this study. Diagnosis was based on clinical suspicion, followed by the detection of GA in the urine, and confirmed on genetic screening for the mutations.
A retrospective review of the inpatient and outpatient electronic hospital records was done. The following parameters were evaluated at diagnosis: age, gender, premorbid medical history, anthropometry, presenting problems, clinical findings and details of management. Anthropometric assessment was done by measuring weight, height/ length and head circumference expressed as percentiles and compared with the reference population. All patients were examined by a paediatric neurologist. Biochemical data (lactate, ammonia and urine organic acid profile) and results of mutational analysis on genetic testing were also collated. All neuroimaging was reviewed by a single radiologist (AM). The magnetic resonance imaging (MRI) brain protocol sequences used included: T1-weighted imaging (T1WI) sagittal, T2-weighted imaging (T2WI) axial, T2WI fluid attenuated inversion recovery (FLAIR) axial, T1WI axial and T2WI coronal, diffusion-weighted imaging (DWI), apparent diffusion coefficient maps and post-contrast T1W1. Information regarding developmental and neurological outcomes was obtained at the last follow-up visit.
Statistical analysis of the data was performed using standard statistical methods in SPSS version 10.0 (SPSS Inc., USA).
Ethics approval for the study was obtained from the University of KwaZulu-Natal Ethics Committee, and institutional approval was obtained from the KwaZulu-Natal Department of Health.
Results
Six patients (4 girls, 2 boys) were identified from our database of 3 500 clinic patients from 2007 to 2015. Therefore, the prevalence of GA1 in our study population was 1.7/1 000 patients. The mean age at diagnosis was 12 (range 7 - 22) months. All patients were of black African indigenous ancestry. Two children had a history of premorbid developmental delay. Four children were diagnosed on presentation with an acute encephalopathic crisis following an intercurrent infection. Five patients had macrocephaly at presentation. Five patients had neuroregression after the acute presentation. The main clinical findings on presentation were: hypotonia (n=4), hemiparesis (n=1) and spasticity (n=1). Four children had seizures during their clinical course. Three had dystonia and 4 had bulbar dysfunction. Four children were treated with antibiotics or antiviral medication during the acute period for suspected underlying meningitis or encephalitis. Long-term therapy included anticonvulsants in 4 children (topiramate in 2 and lamotrigine in 2, respectively) and car-bidopa in 2. All patients were commenced on a lysine-restricted diet, L-carnitine and vitamin Be supplementation. Only 2 patients were able to strictly adhere to the diet.
All patients had elevated glutaryl-carnitine on urine organic acid screening. The A293T mutation is the most commonly described gene mutation in South Africans of black indigenous ancestry.!4' Patients were screened only for the A293T mutation because of the high cost of the full mutational analysis. Five patients showed a homozygous A293T mutation. One patient had a R402W mutation that is not commonly described in black Africans of indigenous ancestry. There was no history of consanguinity in any of our patients.
Three patients underwent computed tomography scans of the brain. Two scans showed frontotemporal atrophy with destruction of the temporal lobes and one scan demonstrated hyperdense basal ganglia.
All patients underwent MRI scans of the brain on presentation (Table 1). All images of patients showed hyperintense basal ganglia on T2WI/FLAIR sequences (Fig. 1). Four children had enlarged basal ganglia and 2 showed atrophy. There was restricted diffusion of the basal ganglia on images of all patients with enlarged basal ganglia. The 2 children presenting without encephalopathic crises had atrophic ganglia. The thalamus was hyperintense in 1 patient. Restricted diffusion of the basal ganglia, cerebral peduncles and central tegmental tract (Fig. 2) was seen on images of all children who presented in encephalopathic crises. Restricted diffusion on DWI images indicates acute cerebral injury. Dysmyelination of the periventricular white matter was seen in 1 patient. Six patients (Fig. 3) had enlarged cerebrospinal fluid (CSF) spaces anterior to the temporal lobes and widened sylvian fissures, while 3 patients had cortical atrophy. Subdural collections were absent in our patients.
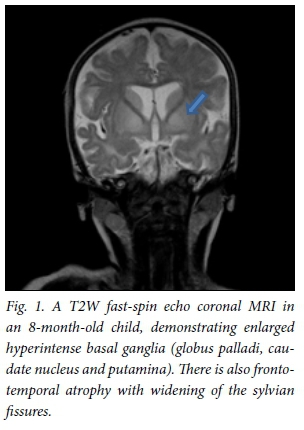
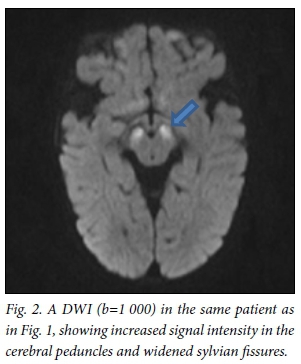
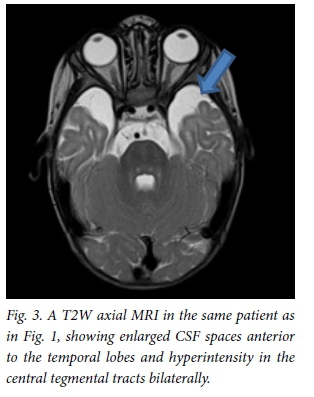
All patients were treated with L-carnitine, multivitamins and a modified diet, with restricted leucine and isoleucine. Families were counselled about the risk of recurrence, prognosis and need for newborn screening in future pregnancies.
On follow-up, 2 patients had a static clinical course with no further regression or gain in milestones, 1 has gained milestones and is now able to sit unaided at 1 year of age. Three children have shown further neuroregression. Of the patients who adhered to the diet, 1 gained developmental milestones and 1 has shown a static clinical course. The mean age at diagnosis of the children who showed progressive disease was higher (16 months) than for those with a non-progressive course (8 months).
Discussion
The estimated prevalence of GA1 is 1/100 000 in newborns.[2,3] Certain populations, such as the Old Order Amish of Pennsylvania, USA,[4] and the Oji-Cree Island Lake First Nation communities of Canada,[5] are known to have a higher disease burden. A higher carrier rate of the A293T mutation has been previously described in patients of black indigenous ancestry in the SA population.[6] Heterozygosity for the A293T mutation was found in 1 of 36 unrelated black SA newborns, giving a predicted prevalence rate for GA1 of 1/5 184 in this population.[6] All patients in our cohort were of black African indigenous ancestry.
There is no newborn screening programme for GA1 in SA. Therefore, diagnosis in our patients was after the newborn period when the patients were already symptomatic. Early presymptomatic diagnosis and focused treatment of GA1 patients lead to a better neurological outcome.
The first presentation of GA1 is usually that of an encephalopathic crisis following an intercurrent infection in infancy,[7] which occurred in 4 of our patients. Our study confirms the findings of a previous study that the earlier the age at first crisis, the higher the risk of neurological morbidity.[8] Neuroregression after an acute presentation is another common clinical manifestation.[9] This was observed in 5 of 6 of our patients. The frequency of macrocephaly (83%) in our patients was higher than that described in the literature.[10] Seizures are an uncommon long-term complication of GA1, which accompanies an acute encephalopathic crisis.[11] Four of our patients had epilepsy. Sodium valproate should not be used for seizure control in patients with GA1, as there have been reports of worsening of the control.[11] L-carnitine is used for the treatment of GA1, as it enhances excretion of GA. Valproic acid reduces the levels of L-carnitine and competes with GA for esterification with L-carnitine. Valproic acid also causes relative L-carnitine deficiency, as it is excreted in the urine as valproylcarnitine. The mechanism of valproate toxicity is purported to be via interference with mitochondrial beta oxidation.[12] Neurotoxicity in GA1 is also mediated through mitochondrial injury. Three of our patients had a movement disorder. Dystonia has been reported to occur frequently in other case series.[13] The severity of dystonia correlates with the extent of striatal injury.[14]
With regard to diet, lysine restriction, carnitine supplementation and aggressive management of intercurrent illness with additional glucose significantly reduce the frequency of acute encephalopathic crises and long-term morbidity.[8] All our patients were commenced on carnitine and multivitamin supplementation. Dietary lysine restriction was initiated in all patients. However, only 2 patients were able to strictly adhere to their diet prescription. The caregivers of the remaining patients declined the prescribed restrictive diet because of cultural preferences.
The most common abnormality seen on MRI as described in the literature is widening of the CSF spaces anterior to the temporal lobes and of the sylvian fissures,[14] which was seen in all our patients.
Acute encephalopathy is typically associated with acute striatal necrosis. Patients with striatal injury develop movement disorders. Our patients showed features of acute injury to the basal ganglia at the time of the acute encephalopathic crises, as evidenced by enlarged basal ganglia with restricted diffusion.
DWI is an MRI sequence used to detect diffusivity of water molecules. Restricted diffusion of extracellular fluid is a result of cytotoxic oedema, which occurs after acute neuronal injury. DWI detects basal ganglia injury earlier and more accurately than conventional MRI sequences.[15] Diffusion-weighted MRI could lead to earlier detection of patients with GA1. Early intervention after detection of acute injury on DWI could result in better long-term outcomes in patients with GA1.
White matter changes involving the deep periventricular white matter are described in patients with GA1.[16] These changes are seen more commonly in patients with late-onset disease. One of our patients showed dysmyelination. Signal change in the central tegmen-tal tract is described in the literature in up to 50% of children and was seen in 66% of our patients.[14] Furthermore, signal changes of T2 hyperintensity and DWI in the corticospinal tracts of the cerebral peduncles were seen in 4 patients who presented in acute encephalopathic crises.
GA1 is a heterogeneous disease. The R402W mutation is the most common mutation in white people.[17] The majority of our patients (83%) tested positive for the A293T mutation, which is the most common mutation in the indigenous black SA population.[6]
Children diagnosed early with GA1 and thereafter started on treatment had better long-term outcomes. Initiation of dietary modification after the onset of symptoms was not effective in preventing permanent neurological injury. However, treatment prevents progressive neurological deterioration.
Practice parameter
GA1 has a high prevalence in SA. Screening for GA1 is mandatory in patients with:
• early-onset macrocephaly and suggestive neuroimaging features of GA1
• neuroregression and dystonia after an acute encephalopathic illness
• unexplained subdural haemorrhage.
• a family history of GA1.
Recommended screening for GA1
This comprises an organic acid profile in urine by means of urine metabolic screening. The diagnosis can be confirmed by testing for the A293T mutation of the GCDH gene, which can be done at the National Health Laboratory Service.
Conclusion
GA1 is an important, treatable neurometabolic disorder with a high prevalence (1.7/1 000) in the SA population. Although the limitations of this study were the small number of patients and that it was retrospective, it is the first study in Africa to analyse the neuroimag-ing correlates in GA1. GA1 is probably being underdiagnosed. In the absence of a newborn screening programme, a high index of clinical suspicion is mandatory for affected children to be identified early. Early diagnosis and implementation of appropriate therapy result in better neurological outcomes.
This study highlights the urgent need for a newborn screening programme to be implemented in SA. Our study also forms the basis for a long-term multisite collaborative study on GA1 in SA.
REFERENCES
1. Stenson PD, Mort M, Ball EV, et al. Human Gene Mutation Database: 2008 update. Genome Med 2009;22(1):13. http://dx.doi.org/10.1186/gm13 [ Links ]
2. Boneh A, Beauchamp M, Humphrey M, et al. Newborn screening for glutaric aciduria type I in Victoria: Treatment and outcome. Mol Genet Metab 2008;94(3):287-291. http://dx.doi.org/10.1016/j.ymgme.2008.03.005 [ Links ]
3. Hedlund GL, Longo N, Pasquali M. Glutaric acidemia type 1. Am J Med Genet C Semin Med Genet 2006;142C(2):86-94. [ Links ]
4. Strauss KA, Lazovic J, Wintermark M, Morton DH. Multimodal imaging of striatal degeneration in Amish patients with glutaryl-CoA dehydrogenase deficiency. Brain 2007;130:1905-1920. http://dx.doi.org/10.1093/brain/awm058 [ Links ]
5. Greenberg CR, Prasad AN, Dilling LA, et al. Outcome of the first 3 years of a DNA-based neonatal screening program for glutaric acidemia type 1 in Manitoba and northwestern Ontario. Canada Mol Genet Metab 2002:75(1):70-78. http://dx.doi.org/10.1006/mgme.2001.3270 [ Links ]
6. Van der Watt G, Owen EP, Berman P, et al Glutaric aciduria type 1 in South Africa - high incidence of glutaryl-CoA dehydrogenase deficiency in black South Africans. Mol Genet Metab 2010;101(2-3):178-182. http://dx.doi.org/10.1016/j.ymgme.2010.07.018 [ Links ]
7. Strauss KA, Puffenberger EG, Robinson DL, et al. Type I glutaric aciduria, part 1: Natural history of 77 patients. Am J Med Genet 2003;121(C):38-52. http://dx.doi.org/10.1002/ajmg.c20007 [ Links ]
8. Bjugstad KB, Goodman SI, Freed CR. Age at symptom onset predicts severity of motor impairment and clinical onset of glutaric aciduria type I. J Pediatr 2000;137(5):681-686. http://dx.doi.org/10.1067/mpd.2000.108954 [ Links ]
9. Hoffmann GH, Zschocke J. Glutaric aciduria type I: From clinical, biochemical and molecular diversity to successful therapy. J Inherit Metab Dis 1999;22(4):381-391. [ Links ]
10. Bijarnia S, Wiley V, Carpenter K, et al Glutaric aciduria type I: Outcome following detection by newborn screening. J Inherit Metab Dis 2008;31(4):503-507. http://dx.doi.org/10.1007/s10545-008-0912-z [ Links ]
11. Mcclelland V, Bakalinova DB, Hendricksz C, Singh RP. Glutaric aciduria type 1 presenting with epilepsy. Dev Med Child Neurol 2009;51(3):235-239. http://dx.doi.org/10.1111/j.1469-8749.2008.03240.x [ Links ]
12. Silva MF, Aires CC, Luis PB, et al. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: A review. J Inherit Metab Dis 2008;31(2):205-216. http://dx.doi.org/10.1007/s10545-008-0841-x [ Links ]
13. Kyllerman M, Skjeldal OH, Lundberg M, et al. Dystonia and dyskinesia in glutaric aciduria type I: Clinical heterogeneity and therapeutic considerations. Mov Disord 1994;9(1):22-30. http://dx.doi.org/10.1002/mds.870090105 [ Links ]
14. Harting I, Neumaier-Probst E, Seitz A, et al. Dynamic changes of striatal and extrastriatal abnormalities in glutaric aciduria type I. Brain 2009;132:1764-1782. http://dx.doi.org/10.1093/brain/awp112 [ Links ]
15. Elster AW. Glutaric aciduria type I: Value of diffusion-weighted magnetic resonance imaging for diagno sing acute striatal necrosis. J Comput Assist Tomogr 2004;28:98-100. http://dx.doi.org/10.1097/00004728- [ Links ]
16. Twomey E, Naughten E, Donoghue V, Ryan S. Neuro-imaging findings in GA1. Pediatr Radiol 2003;33(12):823-830. http://dx.doi.org/10.1007/s00247-003-0956-z [ Links ]
17. Zschocke J, Quak E, Guldberg P, Hoffman GF. Mutation analysis in glutaric aciduria type 1. J Med Genet 2000;37(3):177-181. http://dx.doi.org/10.1136/jmg.37.3.177 [ Links ]
 Correspondence:
Correspondence:
R Govender
govenderr2@ukzn.ac.za
Accepted 10 January 2017


{kind=link}