










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.107 no.2 Pretoria Fev. 2017
http://dx.doi.org/10.7196/samj.2017.v107i2.10849
RESEARCH
Burden, genotype and phenotype profiles of adult patients with sickle cell disease in Cape Town, South Africa
G D PuleI; K MnikaII; M JoubertIII, IV; S MowlaV; N NovitzkyVI, VII, VIII; A WonkamIX
IPhD; Division of Human Genetics, Department of Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
IIBSc Hons; Division of Human Genetics, Department of Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
IIIMB ChB; Haematology Clinic, Groote Schuur Hospital, Cape Town, South Africa
IVMB ChB; National Health Laboratory Service, Groote Schuur Hospital, Cape Town, South Africa
VPhD; Division of Haematology, Departments of Internal Medicine and Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
VIMD, PhD; Haematology Clinic, Groote Schuur Hospital, Cape Town, South Africa
VIIMD, PhD; Division of Haematology, Departments of Internal Medicine and Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
VIIIMD, PhD; National Health Laboratory Service, Groote Schuur Hospital, Cape Town, South Africa
IXMD, DMedSc, PhD; Division of Human Genetics, Department of Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
BACKGROUND. An exponential increase in the number of sickle cell disease (SCD) patients in paediatric services in Cape Town, South Africa, has been reported. The trend in adult/adolescent services has not been investigated.
OBJECTIVES. To evaluate epidemiological trends of SCD and the profile of patients affected by SCD attending the Haematology Clinic at Groote Schuur Hospital (GSH), Cape Town.
METHODS. (i) A retrospective review of the number of SCD patients over the past 20 years; (ii) a cross-sectional analysis of clinical and haematological characteristics of SCD patients; and (iii) molecular analysis of the haemoglobin S mutation, the haplotype in the β-globin-like genes cluster, the 3.7 kb α-thalassaemia gene deletion and 19 selected single-nucleotide polymorphisms (SNPs) associated with fetal haemoglobin (HbF) levels.
RESULTS. From 1995 to 2016, 81 adolescent/adult patients with SCD were registered, mostly originating from other African countries (n=61, 75.3%). There was an increase of over 200% in new cases (n=47) during the last quarter of the two decades investigated. Data from 34 of 58 regular attendees (58.6%) were analysed. The mean age of the patients was 26.1 years (standard deviation (SD) 9.8), and 70.6% were male. With the exception of four patients with sickle/p-thalassaemia, all the patients had SCD (haemoglobin SS). The co-inheritance of a single 3.7 kb α-globin deletion was found in 42.3% of cases (n=11). The Bantu haplotype was the most observed (65.4% of chromosomes). Most HbF-promoting SNPs were not associated with variable levels of haematological indices.
CONCLUSIONS. There is an increasing burden of adult SCD patients at GSH. National health and academic institutions need to adapt policies and healthcare professional training accordingly.
Sickle cell disease (SCD) was the first well-documented molecular disease[1] and is the most prevalent monogenic disease in the world. SCD is an accepted worldwide health problem that is comparable to other major global diseases such as diabetes, hypertension and communicable diseases.[2] Sub-Saharan Africa (SSA) has the highest burden of SCD disease, with in excess of 300 000 new affected births annually, accounting for 80% of all annual affected child births globally.[3] In spite of the high burden of disease in SSA, SCD is often associated with limited to poor medical resources, infrastructure and quality of care, so estimates of neonatal and childhood mortality remain high, with up to 90% of affected children dying by 5 years of age.[1] SCD is caused by the polymerisation and precipitation of the β-globin chains (HbS) during deoxygenation and dehydration of erythrocytes.[5] The altered structure of erythrocytes (normal biconcave shape to a crescent shape) is the basis of the vascular pathology of the disease, which includes abnormality of platelet and leucocyte adhesion and hypercoagulation leading to microvascular occlusion, haemolysis, hypoxia, failed nitric oxide production and multiorgan damage.[5] The hallmark phenotypes of the disease include vaso-occlusive crises, stroke and acute chest syndrome.[5-7] The phenotype of SCD is influenced by both environmental and genetic factors. Variants at three principal loci, BCL11A, HBS1L-MYB intergenic polymorphism and the β-globin haplotype, have been shown to account for 10 - 20% of the variance of fetal haemoglobin (HbF) levels and to be associated with the amelioration of SCD symptoms.[8-10] Other variants in the BCL11A erythroid-specific enhancer (rs1427407 and rs7606173) have been shown to account for 8% and 6.2% of HbF variance, respectively, among SCD patient cohorts in the USA,[11,12] Tanzania[13] and Cameroon.[4] The co-inheritance of α-thalassaemia has also been associated with improved clinical manifestations of SCD.[15-18] Although the multiple independent origins of the HbS mutation have been questioned recently,[19] the SCD mutation is classically associated with five region-defined β-globin gene haplotypes, Benin, Bantu or Central African (CAR), Cameroon, Senegal and Indian-Arab,[20-23] four of which are from Africa and associated with malaria incidence.[24]
Because of the low incidence of malaria, the incidence of SCD in South Africa (SA) is equally extremely low; the HbS allele can be found in some indigenous SA ethnic groups (Venda and Shangaan) at an approximated frequency of 0.2%.[25-26] However, this is changing with the socioeconomically motivated influx of immigrants from other African countries, especially those within the equatorial malaria-endemic belt, resulting in a 300 - 400% increase in new cases of SCD over the past 10 years at Red Cross War Memorial Children's Hospital (RCWMCH) in Cape Town, SA.[27] The existence of similar trends in adult SCD patient services has not been investigated. Following our previous report at RCWMCH, we report in the present study the trend of new cases of adolescent and adult SCD over the past 20 years, having studied the clinical, haematological and genetic profiles of a cohort of 34 adolescent and adult SCD patients at the Haematology Unit at Groote Schuur Hospital (GSH), Cape Town.
Methods
Ethical approval
The study was performed in accordance with the Declaration of Helsinki and with the approval of the Faculty of Health Sciences Human Research Ethics Committee, University of Cape Town (HREC ref. no. 132/2010). Informed and written consent was obtained from adult participants (>18 years), and for one patient aged 15 years informed consent was obtained from the guardian with assent from the participant.
Patients
The Haematology Clinic runs weekly every Wednesday. Most patients are seen at least once a month, and clinically stable patients every 3 months, with the exception of crisis-related hospitalisation. A retrospective review of the number of SCD patients attending the clinic over the past 20 years and a cross-sectional analysis of patients who regularly attend the clinic were performed. Clinical events and haematological indices were retrospectively collected from hospital records. The haematological measures were those reported at the first visit to the hospital.
Molecular methods DNA extraction
DNA was isolated from the peripheral blood using the AllPrep DNA/RNA/miRNA universal kit (Qiagen, USA) according to the manufacturer's instructions.
Genotyping
HbS mutation and β-globin haplotypes
Polymerase chain reaction (PCR) and Ddel restriction analysis were used to confirm the presence of the HbS mutation using 100 ng DNA. [28] Published primers and methods[29] genotyping five restriction fragment length polymorphic regions in the β-globin gene cluster were used to analyse XmnI (5Όγ), HindlII (Gy), HindlII (Ay), HinclI (3''Ψβ) and HinfI (5'β) to determine the β-globin haplotype background.[19]
Single-nucleotide polymorphisms (SNPs)
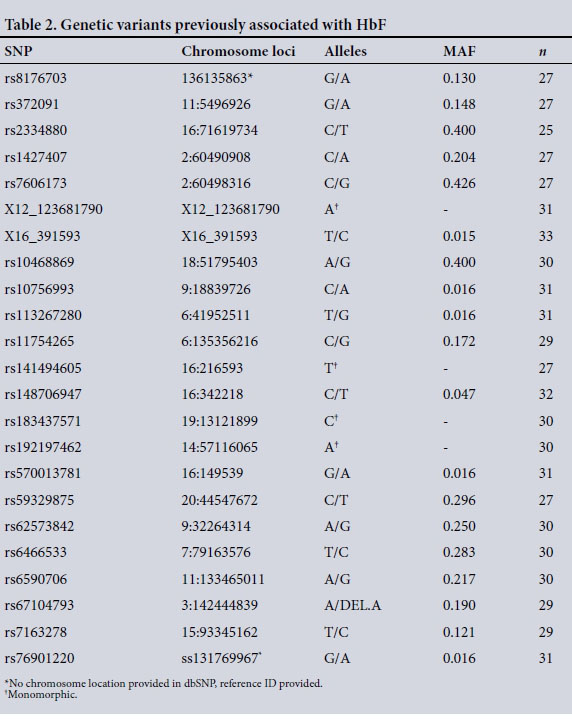
Using a reported method,[9] SNaPshot genotyping, capillary electrophoresis and direct cycle sequencing were used to assay five selected HbF-associated variants: rs8176703, rs372091, rs2334880, rs1427407 and rs7606173. In addition, 18 other variants were analysed using the iPLEX Gold Sequenom Mass Genotyping Array (Inqaba Biotec, SA): X12_123681790, X16_391593, rs10468869, rs10756993, rs113267280, rs11754265, rs141494605, rs148706947, rs183437571, rs192197462, rs570013781, rs59329875, rs62573842, rs6466533, rs6590706, rs67104793, rs7163278 and rs76901220.
Statistical analysis
Descriptive statistics were obtained for all quantitative data using SPSS version 21.0 (IBM, USA). A χ2 test with one degree of freedom was used to perform the Hardy-Weinberg Equilibrium (HWE) test on the SNP genotypes with all variants in HWE (p>0.05).
Results
Patients' origin and trends
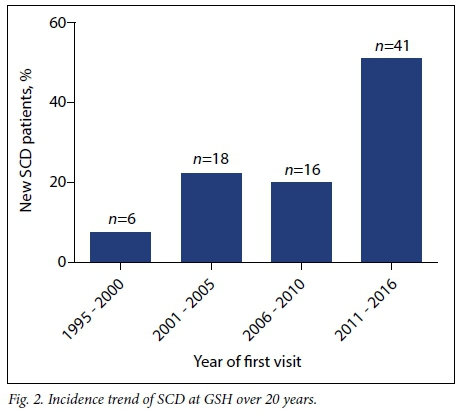
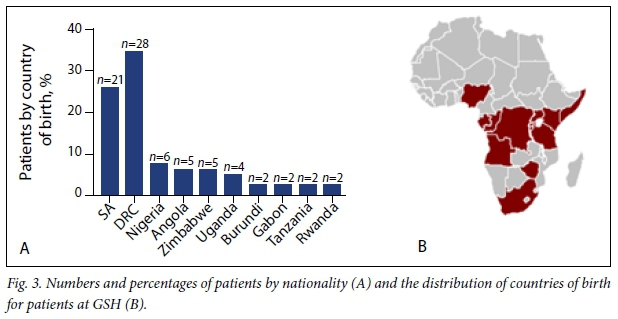
A total of 128 patients' files from 1995 to March 2016 were reviewed. Among them, 47 patients were diagnosed with some form of α- or β-thalassaemia (Fig. 1). Of the remaining 81 patients affected by SCD, 61 (75.3%) were from other SSA countries. Over the last quarter (2011 - 2016) of the past two decades, there was an approximately 200% increase in new cases of SCD registered at the GSH Haema-tology Clinic (n=47) (Fig. 2). Fig. 3 shows the number of patients seen at GSH (A) and countries of origin (B), with 16.4% (n=21) SA patients, most of whom are of mixed/Indian ancestry (n=15), and 21.9% (n=28) from the Democratic Republic of Congo (DRC).
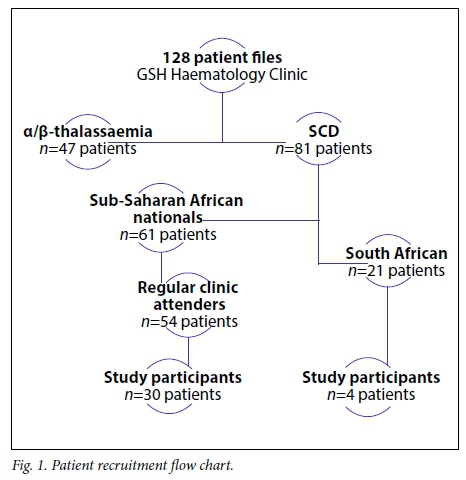
Of the 58 patients who regularly attend the Haematology Clinic, 34 (58.6%) consented to inclusion in the study (Fig. 1). Over 75% of the patients at GSH were referrals from RCWMCH, with others coming from neighbouring secondary-level hospitals in Cape Town and a minority of internal referrals of relatives of attending patients.
Clinical and haematological profile
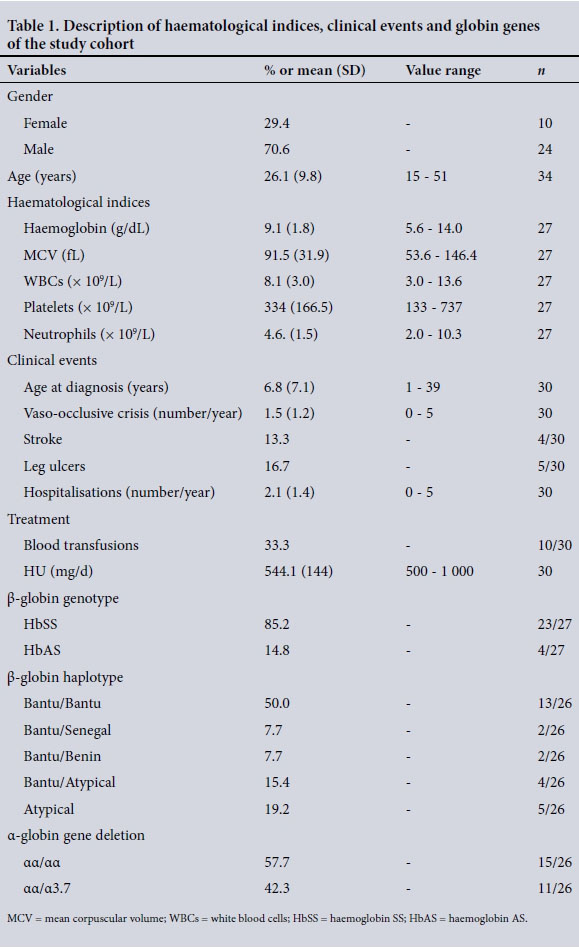
The mean age was 26.1 years (standard deviation (SD) 9.8, range 15 -51), and 70.6% were male. The rate of co-inheritance of a single 3.7 kb α-globin gene deletion was 42.3% (n=11).
Table 1 summarises the haematological and clinical events recorded for all patients. Information obtained from the anamnesis indicated that the majority of the patients were diagnosed relatively late (mean age at diagnosis 6.8 years (SD 7.1), range 1 -39), as a result of the presentation of the initial clinical manifestations of SCD, mainly pain episodes.
Clinical management
With regard to treatment, 33.3% (n=10) of the patients in the cohort had received at least one blood transfusion. About 16.7% (n=5) of the patients had been enrolled in a hypertransfusion programme, ranging from a fortnightly to monthly transfusion regimen, to manage complications such as stroke, chronic pain crises and non-healing chronic leg ulcers. The frequency of transfusions is largely dependent on symptom severity and availability of blood units from the Western Cape Blood Transfusion Service, Cape Town, SA.
Of the patient cohort, 86.6% (n=26) were at maximum tolerated dose of hydroxyurea (HU) with dosages ranging between 500 and 1 000 mg/d. From our patient survey, 30 - 50% of the participants were fully compliant with HU treatment, 20 - 30% reported partial compliance (tending to forget to take the treatment two to three times a week), and some patients refused treatment. Reasons for refusal included potential cancer development in the future, family planning, particularly for men afraid of treatment-related infertility, and self-perceived improvement of symptoms without the treatment.
Genetic characteristics
Sickle cell genotypes: β-globin haplotypes and co-inheritance of α-thalassaemia
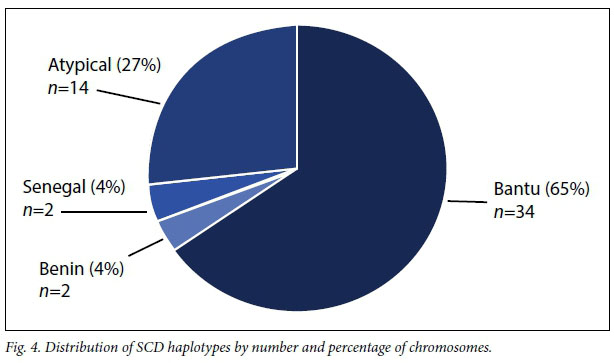
The description of the HbS allele frequency, β-globin haplotype background and α-globin gene deletion for the patients is given in Table 1. Genotyping for the HbS mutation revealed that 85.2% (n=23) of the patients were homozygous for the mutation (haemoglobin SS), with the rest being heterozygous (haemoglobin AS) with a possibility of β0-thalassaemia (HbS/β0) (n=4), all of whom were South African with mixed and Indian ancestry. Fig. 4 shows the distribution of the SCD β-globin gene haplotypes: the Bantu and Atypical haplotypes accounted for 65.4% and 26.9%, respectively, whereas the Senegal and Benin haplotypes accounted for 3.8% each, with no observation of the Cameroon and Indian-Arab haplotypes. In combination, the Bantu/Bantu haplotype represented 50.0% of the patients (Table 1). The heterozygous 3.7 kb α-globin gene deletion was observed in 42.3% of the patients.
Frequency of genetic variants associated with HbF levels
Table 2 shows the observed alleles and minor allele frequency (MAF) of genetic variants previously associated with HbF, some recently in a Sardinian population. [30] All variants were in HWE (p>0.05) with the exception of four loci (X12_123681790, rs141494605, rs183437571 and rs192197462) that presented monomorphic alleles in all patients. Tests of association between the variants and all haematological indices including Hb levels were conducted; however, no significant associations were observed except for rs6466533 and the SCD haplotype combinations. The CC genotype (rs6466533) was associated with higher platelet counts than the heterozygous TC genotype (p<0.05).
Similarly, the Bantu/Bantu haplotype combination was associated with higher platelet counts than the Atypical/Atypical genotype (p<0.05). Table 3 shows the MAF of the above variants in African populations: Esan (Nigeria), Luhya (Kenya), Mandinka (Gambia) and Mende (Sierra Leone); American (including African-American), European and both East and South Asian populations.
Discussion
To the best of our knowledge, this is the first study describing the clinical and genetic backgrounds of SCD patients at GSH and reporting on adult patients with SCD in SA. The results of this study indicate a similar trend of a rapid increase in the number of cases of SCD that was previously reported at RCWMCH in Cape Town.[27] This was also the result of migration from SSA countries where SCD is most prevalent. Related to this was a specific administrative difficulty in taking care of some patients who lack the up-to-date and correct paperwork for immigrants and asylum seekers. This was an indirect indication that most patients arrived as adults in SA, contrary to the observation in the second part of the last decade at RCWMCH, where most patients were SA born. It is therefore expected that the adult SCD population at GSH will continue to grow from the compounded effects of future referrals from neighbouring paediatric hospitals and the arrival of new adult patients from migrant populations, as migration, particularly into SSA, continues to be the reality for many people seeking political asylum, economic opportunities and better healthcare. Concomitant with this migration, the improved clinical management and healthcare of paediatric SCD patients is expected to increase the pool of adult patients living with SCD. This will increase the number of patients who will survive well beyond reproductive age, which is likely to increase the frequency of the HbS allele in the population.
Newborn screening and comprehensive clinical care programmes, which are also possible in SA, have reduced SCD-related premature childhood deaths by 70% in high-income nations such as the USA,[31,32] and most patients can survive into adulthood. [33] A similar increasing trend of SCD in countries previously not affected by the disease has been observed in Ireland,[34] Italy,[35] Germany,[36] England[37] and France.[38] Therefore, the evidence that the SCD burden is comparable to that of communicable diseases and other major global diseases such as hypertension and diabetes[2] will have increasing resonance. The marked increase in patients between 2001 and 2005 could also be associated with the ending of a civil war, a transitional government and political instability in DRC, the effects of which had spread into neighbouring states. The more recent increase (2011 - 2016) is more likely to be due to economic and health-motivated migration. The increase of SCD in both paediatric and adult settings will impose a new burden on the healthcare system in SA concomitant with a new need for training at all levels of medical education, as well as the need for policies from health authorities for the prevention, management and care of haemoglobinopathies.
The number of annual vaso-occlusive crises was similar to that reported in Cameroon, as was the mean number of vaso-occlusive crises per year.[9] Major challenges faced by healthcare professionals at GSH were patient compliance with HU treatment, compliance with supportive medication such as folic acid and patient clinic attendance. There were several barriers to HU treatment, including the financial implications of taking time off work to attend the clinic and receive it (maximum one month's supply), and misconceptions about the treatment and its possible carcinogenic effects, as well as potential impotence in male patients.[39] Most patients who fail to comply with clinic appointments do so simply because they feel better and therefore see no need to go to the hospital.
The novel aspect of this article is the report on the genetic background of four key modifiers of HbF: variants at the BCL11A erythroid-specific enhancer, β-globin haplotypes, α-thalassaemia 3.7 kb gene deletion and several other known HbF-promoting polymorphisms. It is imperative to gain a better understanding of genetic variants affecting the predisposition to specific complications such as stroke and acute chest syndrome, and polymorphisms affecting susceptibility to pain, as well as the pharmacogenomics of commonly prescribed treatments such as HU, malaria prophylaxis and pain medication. The dominance of the Bantu haplotype in this cohort is in accordance with the Congolese origin of most patients. Indeed, the Bantu haplotype is most prevalent below the equatorial malaria belt across southern African countries.[19] Most variants were not associated with haematological indices except the CC genotype at rs6466533 and Bantu/Bantu haplotype combination with platelet counts, probably because of the modest sample size. The differences in MAF of the recently identified HbF-promoting loci in a Sardinian population[30] among African populations from the Human 1000 Genome Project (1000G) (Table 3) emphasises the necessity of large-scale genomic analyses on various populations across the continent, as there are vast variations between any two African populations.
It should be made clear that the intention of this article is not to stigmatise SCD nor immigrant patients, but to inform and prepare medical care providers and healthcare officials of the increasing need for management of haemoglobinopathies in SA. This trend is not restricted to SA, with countries such as Italy[40] the Republic of Ireland,[34] England[41] and Germany[36] affected by the reality of population movement and new burden of disease, developing neonatal screening programmes and establishing SCD centres in response to similar increases in SCD prevalence.
Study limitations
The final sample size of patients included in the present study was limited because of poor patient compliance with clinic attendance, self-transfers to other hospitals and the shortened period of recruitment. The sample size did not allow for robust statistical analyses to reveal potent markers of specific phenotypes or clinical measures. HbF levels were measured for a handful of patients performed using high performance liquid chromatography before initiation of HU treatment. This haematological measure would have been ideal to check for association with genetic variants as baseline HbF.
Conclusions
Over the past 10 years, the number of adult patients living with SCD has increased considerably, imposing the creation of a weekly outpatient service at GSH. The genetic profile is similar to that of many other SCD patients from the other SSA countries from where most patients originate. The trend has a number of implications, particularly for medical education at academic and training institutions, policy action on prevention and care at the National Department of Health, and research in haemoglobinopathies in SA.
Acknowledgments. We would like to thank the nursing staff of the Haematology Service at GSH for their support and the patients for their participation.
Contribution to authorship. GP and AW conceived and designed the experiments. GP, KM and MJ recruited and sampled the patients. GP and KM performed the experiments. GP, KM and AW analysed the data. AW, MJ, SM and NN contributed reagents/materials/analysis tools. GP and AW wrote the article. GP, KM, MJ, SM, NN and AW revised and approved the manuscript.
Funding statement. The molecular experiments of the study were funded by the National Health Laboratory Services, SA and the National Institutes of Health, USA, grant no. 1U01HG007459-01. The students' bursaries were supported by the Oppenheimer Memorial Trust, the National Research Foundation, and FirstRand Laurie Dippenaar Scholarship, SA. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
1. Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910. Yale J Biol Med 2001;74(3):179-184. http://dx.doi.org/10.1001/archinte.1910.00050330050003 [ Links ]
2. Weatherall DJ. Hemoglobinopathies worldwide: Present and future. Curr Mol Med 2008;8(7):592-599. http://dx.doi.org/10.2174/156652408786241375 [ Links ]
3. Piel FB, Patil, AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: A contemporary geostatistical model-based map and population estimates. Lancet 2013;381(9861):142-151. http://dx.doi.org/10.1016/S0140-6736(12)61229-X [ Links ]
4. Makani J, Menzel S, Nkya S, et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood 2011;117(4):1390-1392. http://dx.doi.org/10.1182/blood-2010-08-302703 [ Links ]
5. Bartolucci P, Galacteros F. Clinical management of adult sickle-cell disease. Curr Opin Hematol 2012;19(3):149-155. http://dx.doi.org/10.1097/MOH.0b013e328351c35f [ Links ]
6. Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010;115(22):4331-4336. http://dx.doi.org/10.1182/blood-2010-01-251348 [ Links ]
7. Malowany JI, Butany J. Pathology of sickle cell disease. Semin Diagn Pathol 2012;29(1):49-55. http://dx.doi.org/10.1053/j.semdp.2011.07.005 [ Links ]
8. Thein SL, Menzel S, Peng X, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci USA 2007;104(27):11346-11351. http://dx.doi.org/10.1073/pnas.0611393104 [ Links ]
9. Wonkam A, Bitoungui VJN, Vorster AA, et al. Association of variants at BCL11A and HBS1L- MYB with hemoglobin F and hospitalization rates among sickle cell patients in Cameroon. PLoS One 2014;9(3):e92506. http://dx.doi.org/10.1371/journal.pone.0092506 [ Links ]
10. Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA 2008;105(33):11869-11874. http://dx.doi.org/10.1073/pnas.0804799105 [ Links ]
11. Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013;342(6155):253-257. http://dx.doi.org/10.1126/science.1242088 [ Links ]
12. Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 2010;42(12):1049-1051. http://dx.doi.org/10.1038/ng.707 [ Links ]
13. Mtatiro SN, Mgaya J, Singh T, et al. Genetic association of fetal-hemoglobin levels in individuals with sickle cell disease in Tanzania maps to conserved regulatory elements within the MYB core enhancer. BMC Med Genet 2015;16:1. http://dx.doi.org/10.1186/s12881-015-0144-7 [ Links ]
14. Pule GD, Ngo Bitoungui VJ, Chetcha Chemegni B, et al. Association between variants at BCL11A erythroid-specific enhancer and fetal hemoglobin levels among sickle cell disease patients in Cameroon: Implications for future therapeutic interventions. OMICS 2015;19(10):627-631. http://dx.doi.org/10.1089/omi.2015.0124 [ Links ]
15. Hsu LL, Miller ST, Wright E, et al. Alpha thalassemia is associated with decreased risk of abnormal transcranial Doppler ultrasonography in children with sickle cell anemia. J Pediatr Hematol Oncol 2003;25(8):622-628. http://dx.doi.org/10.1097/00043426-200308000-00007 [ Links ]
16. Rumaney MB, Bitoungui VJN, Vorster AA, et al. The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS One 2014;9(6):e100516. http://dx.doi.org/10.1371/journal.pone.0100516 [ Links ]
17. Saleh-Gohari N, Mohammadi-Anaie M. Co-inheritance of sickle cell trait and thalassemia mutations in south central Iran. Iran J Public Health 2012;41(10):81-86. [ Links ]
18. Wambua S, Mwacharo J, Uyoga S, et al. Co-inheritance of a+-thalassaemia and sickle trait results in specific effects on haematological parameters. Br J Haematol 2006;133(2):206-209. http://dx.doi.org/10.1111/j.1365-2141.2006.06006.x [ Links ]
19. Bitoungui VJN, Pule GD, Hanchard N, et al. Beta-globin gene haplotypes among Cameroonians and review of the global distribution: Is there a case for a single sickle mutation origin in Africa? OMICS 2015;19(3):171-179. http://dx.doi.org/10.1089/omi.2014.0134 [ Links ]
20. Flint J, Harding, RM, Boyce, AJ, et al. The population genetics of the haemoglobinopathies. Bailliere Clin Hematol 1998;11(1):1-51. http://dx.doi.org/10.1016/s0950-3536(98)80069-3 [ Links ]
21. Elion J, Berg PE, Lapoumeroulie C, et al. DNA sequence variation in a negative control region 5' to the beta-globin gene correlates with the phenotypic expression of the beta s mutation. Blood 1992;79(3):787-792. [ Links ]
22. Pagnier J, Mears JG, Dunda-Belkhodja O, et al. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci USA 1984;81(6):1771-1773. http://dx.doi.org/10.1073/pnas.81.6.1771 [ Links ]
23. Labie D, Pagnier J, Lapoumeroulie C, et al. Common haplotype dependency of high G gamma- globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients. Proc Natl Acad Sci USA 1985;82(7):2111-2114. http://dx.doi.org/10.1073/pnas.82.7.2111_ [ Links ]
24. Williams TN, Mwangi TW, Wambua S, et al. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J Infect Dis 2005;192(1):178-186. http://dx.doi.org/10.1086/430744 [ Links ]
25. Bonafede RP, Botha MC, Beighton P. Inherited anaemias in the Greek community of Cape Town. J Med Genet 1979;16(3):197-200. https://dx.doi.org/10.1136/jmg.16.3.197 [ Links ]
26. Beighton P, Botha M. Inherited disorders in the black population of southern Africa. S Afr Med J 1986;69(4):247-249. [ Links ]
27. Wonkam A, Ponde C, Nicholson N, et al. The burden of sickle cell disease in Cape Town. S Afr Med J 2012;102(9):752-754. http://dx.doi.org/10.7196SAMJ.5886 [ Links ]
28. Saiki RK, Scharf S, Faloona F, et al. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985;230(4732):1350-1354. http://dx.doi.org/10.1126/science.2999980 [ Links ]
29. Steinberg M, Lu Z, Nagel R, et al. Hematological effects of atypical and Cameroon β-globin gene haplotypes in adult sickle cell anemia. Am J Hematol 1998;59(2):121-126. http://dx.doi.org/10.1002/(sici)1096-8652(199810)59:2<121::aid-ajh4>3.0.co;2-# [ Links ]
30. Danjou F, Zoledziewska M, Sidore C, et al. Genome-wide association analyses based on whole- genome sequencing in Sardinia provide insights into regulation of hemoglobin levels. Nat Genet 2015;47(11):1264-1271. http://dx.doi.org/10.1038/ng.3307 [ Links ]
31. Yanni E, Grosse SD, Yang Q, et al. Trends in pediatric sickle cell disease-related mortality in the United States, 1983-2002. J Pediatr 2009;154(4):541-545. http://dx.doi.org/10.1016/j.jpeds.2008.09.052 [ Links ]
32. Vichinsky EP. Comprehensive care in sickle cell disease: Its impact on morbidity and mortality. Semin Hematol 1991;28(3):220-226. [ Links ]
33. Chaturvedi S, DeBaun MR. Evolution of sickle cell disease from a life-threatening disease of children to a chronic disease of adults: The last 40 years. Am J Hematol 2016;91(1):5-14. http://dx.doi.org/10.1002/ajh.24235 [ Links ]
34. Gibbons C, Geoghegan R, Conroy H, et al. Sickle cell disease: Time for a targeted neonatal screening programme. Ir Med J 2015;108(2):43-45. [ Links ]
35. Colombatti R, Perrotta S, Samperi,P, et al. Organizing national responses for rare blood disorders: The Italian experience with sickle cell disease in childhood. Orphanet J Rare Diseases 2013;8:169. http://dx.doi.org/10.1186/1750-1172-8-169 [ Links ]
36. Zur B. Increase in genetically determined anemia as a result of migration in Germany. Internist (Berl) 2016;57(5):444-451. http://dx.doi.org/10.1007/s00108-016-0030-1 [ Links ]
37. Pizzo E, Laverty AA, Phekoo KJ, et al. A retrospective analysis of the cost of hospitalizations for sickle cell disease with crisis in England, 2010/11. J Public Health (Oxf) 2015;37(3):529-539. http://dx.doi.org/10.1093/pubmed/fdu026 [ Links ]
38. Habibi A, Arlet JB, Stankovic K, et al. French guidelines for the management of adult sickle cell disease: 2015 update. Rev Med Interne 2015;36(5 Suppl 1):5S3-S84. http://dx.doi.org/10.1016/S0248-8663(15)60002-9 [ Links ]
39. Pule GD, Mowla S, Novitzky N, et aL A systematic review of known mechanisms of hydroxyurea- induced fetal hemoglobin for treatment of sickle cell disease. OMICS 2015;8(5):669-679. http://dx.doi.org/10.1586/17474086.2015.1078235 [ Links ]
40. Colombatti R, Montanaro M, Guasti F, et al. Comprehensive care for sickle cell disease immigrant patients: A reproducible model achieving high adherence to minimum standards of care. Paed Blood Cancer 2012;59(7):1275-1279. http://dx.doi.org/10.1002/pbc.24110 [ Links ]
41. Daniel YA, Henthorn J. Newborn screening for sickling and other haemoglobin disorders using tandem mass spectrometry: A pilot study of methodology in laboratories in England. J Med Screen 2016;23(4):175-178. http://dx.doi.org/10.1177/0969141316631008 [ Links ]
 Correspondence:
Correspondence:
A Wonkam
ambroise.wonkam@uct.ac.za
Accepted 10 June 2016


{kind=link}
{kind=link}
{kind=link}