










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.107 n.2 Pretoria Feb. 2017
http://dx.doi.org/10.7196/samj.2017.v107i2.12022
RESEARCH
Analysis of mutations causing familial hypercholesterolaemia in black South African patients of different ancestry
U K IbeI; R WhittallI; S E HumphriesII; G PilcherIII; F RaalIV
IBSc, MSc;Centre for Cardiovascular Genetics, British Heart Foundation Laboratories, Institute of Cardiovascular Sciences, University College London, UK
IIBSc, PhD; Centre for Cardiovascular Genetics, British Heart Foundation Laboratories, Institute of Cardiovascular Sciences, University College London, UK
IIIBSc, MSc; Carbohydrate and Lipid Metabolism Research Unit, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVMMed, PhD; Carbohydrate and Lipid Metabolism Research Unit, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND. Familial hypercholesterolaemia (FH) is usually caused by mutations in three genes (LDLR, APOB and PCSK9).
OBJECTIVE. To identify the spectrum of FH-causing mutations in black South African (SA) patients.
METHODS. DNA samples of 16 unrelated South African FH patients with elevated low-density lipoprotein cholesterol levels, tendon xanthomas and corneal arcus (3 clinically homozygous FH and 13 heterozygous FH) of ethnic African origin were screened for mutations in the LDLR (coding region, promoter and intron/exon boundaries), APOB (part of exon 26) and PCSK9 genes (exon 7), using high-resolution melting.
RESULTS. Eight LDLR mutations were identified, for an overall detection rate of 8/19 predicted FH-causing alleles (42.1%). The previously reported six base pair deletion p.(D47_G48del) was found in two patients, and two novel variants (c.1187-25T>C and c.1664T>G p.(L555R)) were found, both predicted to be pathogenic using in silico web-based predictive algorithms. No pathogenic variants in APOB or PCSK9 were found.
CONCLUSIONS. These findings contribute to the knowledge of allelic heterogeneity in the spectrum of FH-causing mutations in black SA patients, signifying their ancestral diversity. The relatively low overall detection rate may reflect locus heterogeneity of the FH phenotype in black SA FH patients.
Familial hypercholesterolaemia (FH; OMIM #143890) is a disorder of lipid metabolism inherited in an autosomal dominant form and characterised by elevated levels of serum cholesterol.[1] Untreated individuals with FH have an increased risk of developing early coronary heart disease (CHD), with a >50% cumulative risk of fatal or non-fatal CHD by age 50 years in men and at least 30% risk in women aged 60 years.[2] The heterozygous form of the disease (HeFH) is thought to occur with a frequency of about 1 in 500 in the population, with the homozygous form (HoFH) occurring with a frequency of 1 in 1 000 000,[3] although recent data in European populations indicate that the frequency may be higher.[4] The prevalence of HeFH in black South African (SA) populations is essentially unknown. FH is caused mainly by mutations in the low-density lipoprotein receptor (LDLR) (OMIM #606945), apolipoprotein B (APOB) (OMIM #107730) and protein convertase subtilisin/kexin type 9 (PCSK9) (OMIM #607786) genes, which are involved in the regulation of plasma lipid levels. Mutations in the LDLR account for the vast majority of the mutations identified in FH patients,[2] and recent studies report >1 200 different LDLR mutations worldwide.[5] Mutations in the LDLR gene result in the impaired uptake or processing of low-density lipoprotein (LDL) particles, which results in increased levels of serum LDL cholesterol (LDL-C), promoting the development of atheroma and increasing the risk of CHD. The LDL particle contains one molecule of apolipoprotein B, which is the ligand for the LDL receptor (LDL-R). In carriers of the mutant APOB, p.(R3527Q), the affinity of LDL-R for R3527Q-LDL is markedly reduced, resulting in the FH phenotype and increased risk of CHD.[6] This mutation explains ~5% of cases of FH in many European countries,[6] but has not been found in subjects of African origin. PCSK9 encodes an enzyme that is involved in the post-translational regulation of LDLR by degrading the LDLR in the lysosome of the cell, preventing its recycling.[7] Gain-of-function mutations in this gene such as p.(D374Y) lead to increased degradation of the LDLRs, and severe hypercholesterolaemia.[8,9] The frequency of this mutation is very low, however, and reported to be <2% in patients of European descent with a clinical diagnosis of FH.[6]
There is a high prevalence of FH due to founder effect in certain isolated populations such as SA Ashkenazi Jews (1 in 72), Lebanese Christians (1 in 85), Afrikaners (1 in 100), Tunisians (1 in 165), and French Canadians (1 in 270).[3,10] In such populations there is a high prevalence of a few mutations causing a large percentage of the FH cases observed. For example, three founder mutations, p.(D227E), p.(V429M) and p.(D175N), account for ~90% of FH cases among the Afrikaners in SA.[11]
There is great value in characterising the FH mutation spectrum of a particular population for population screening in order to identify potential patients at risk by genetic cascade testing and reduce cardiovascular morbidity and mortality. For over a decade, there have been no published findings examining the spectrum of mutations causing FH in the black SA population. We describe findings in 16 such patients with a clinical diagnosis of FH.
Methods
Patient samples and DNA extraction
Blood samples from 16 black SA FH patients of different ancestry attending the lipid clinic in the Department of Medicine, University of the Witwatersrand, Johannesburg, SA, were collected and sent to the Centre for Cardiovascular Genetics, British Heart Foundation Laboratories, University College London. The patients consisted of 3 probands with a clinical diagnosis suggestive of homozygous FH and 13 with heterozygous FH. Ethical approval was obtained from the Charlotte Maxeke Johannesburg Academic Hospital Human Research Ethics Committee (ref. no. M160142), and informed consent was also obtained from patients or their representatives. Genomic DNA was isolated from buffy coat samples using an adaptation of the salting-out method described by Miller et al.[12]
High-resolution melting (HRM) and polymerase chain reaction (PCR)
The promoter (up to position -298) and coding regions of the LDLR gene together with the p.(R3527Q) mutation (part of exon 26) of APOB and p.(D374Y) (exon 7) of the PCSK9 gene were screened by HRM. Designed oligonucleotide primers for HRM-PCR were used to amplify intron-exon junctions and up to 60 base pairs (bp) of the introns of each fragment. The primers used were as described by Whittall et al.[13] All the HRM runs were performed using the Quanta Accumelt kit (Quanta Biosciences, USA) according to the manufacturer's instructions on a Corbett Rotor-Gene6000 system (Corbett Life Science, Australia). Owing to their large sizes, exon 4 (381 bp) of the LDLR gene was screened by four overlapping PCR fragments and exon 10 (228 bp) by two overlapping fragments. Amplification of most of the 3' fragment of exon 4, designated as e4.4, was performed on the G-Storm GS4 thermal cycler (Gene Technologies, UK), as previously described.[13] Used as control samples were known mutation-positive samples and polymorphic samples, which were run alongside patient samples to aid detection of potential mutations in unknown samples, as shown previously.[13] Common variants were detected by PCR and restriction enzyme digestion, as described by Whittall et al.[13]Characteristic HRM profiles of identified variants, difference graph (inset) and annotated sequence profile depicting identified variation are shown in supplementary Fig. 1 (all supplementary figures and tables are available on request from the corresponding author).
Sequencing of PCR fragments
Potential mutant samples revealed by aberrant HRM profiles or discord in restriction fragment length polymorphism bands were purified using the Illustra GFX PCR DNA and Gel Band Purification Kit (both from GE Healthcare Life Sciences, UK), according to the manufacturer's instructions. Samples were sent to Source BioScience LifeSciences, Cambridge, UK, for sequencing. Sequence data were viewed using the freely downloadable FinchTV version 1.4.0 software resource (Geospiza, USA).
Multiplex ligation-dependent probe amplification
Large rearrangements, gross deletions/insertions, copy number variations and aberrations were assayed by multiplex ligation probe amplification using Kit P062 (MRC-Holland, Netherlands), according to the manufacturer's instructions.
Bioinformatics analysis
The pathogenicity of all identified variants was assessed using open-access in silico web-based tools. For changes in amino acids, we used polymorphism phenotyping (polyPhen; http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (SIFT; http://sift.bii.a-star.edu.sg/) and Have yOur Protein Explained (http://www.cmbi.ru.nl/hope/). For variants with potential impact on pre-mRNA splicing, we used Berkeley Drosophila Genome Project Splice Site Prediction by Neural Network (http://www.fruitfly.org/seq_tools/splice.html) and Human Splicing Finder version 2.4.1 (http://www.umd.be/HSF/).
Statistical analysis
All statistical analyses were done using R software for statistical computing (v 3.0.1, 2013; R Foundation, Austria) and Stata 12 (Stata-Corp, USA). Testing for significance of heterogeneity in mutation frequencies in mutation-positive and negative groups was based on the χ2 and Fisher's exact tests. Statistical significance was set at a level of p<0.05.
Results
Subjects screened in this study were clinically diagnosed FH patients, usually with early-onset coronary artery disease (CAD) and/or the same manifestation in a first- or second-degree relative. Three of the 16 probands had a clinical presentation of HoFH and 13 a clinical presentation of HeFH (Table 1); 10 of the probands presented with the clinical signs of tendon xanthoma, all had corneal arcus, and 4 had established CAD.
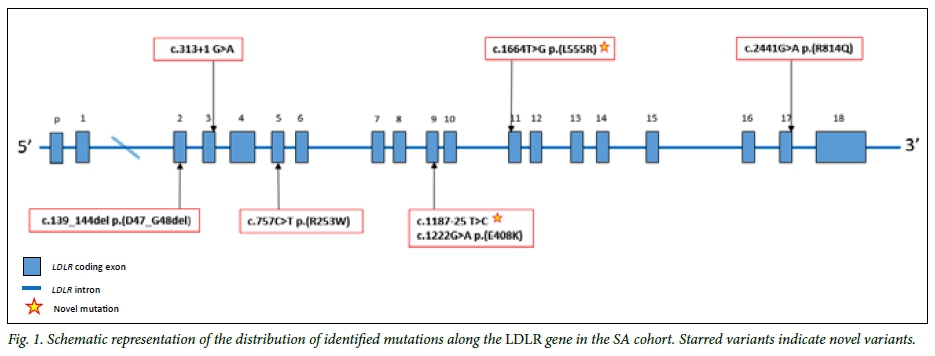
All subjects were screened for mutations causing FH in the LDLR gene (coding region, promoter and intron/exon boundaries), the APOB gene (part of exon 26) and the PCSK9 gene (exon 7). As shown in supplementary Fig. 1, sequencing of the aberrant profiles observed in the HRM analysis resulted in the identification of seven different LDLR variants with the 6 bp deletion seen in two subjects. This 6 bp deletion (FH Cape Town-1), which was first reported in a Xhosa FH homozygote,[14] was seen in patients 1 and 5, of Xhosa and Tswana origin, respectively. The in-frame deletion of 6 bp in the 3' half of exon 2, which encodes the first cysteine-rich repeat, results in the elimination of two amino acids, aspartic acid and glycine (p.(D47_ G48del), from the LDLR and is clearly pathogenic. The identified LDLR mutations in this study cohort is presented in a schematic plot in Fig. 1, with further details shown in supplementary Table 1.
Of the remaining mutations, one missense mutation, p.(L555R), and a splice variant, c.1187-25 C>T at the intron 8/exon 9 junction (Table 2), have not been reported previously in the SA FH population or elsewhere. Patient 10 carried a c.1664 T>G base change, resulting in leucine-to-arginine substitution at position 555 of the LDLR protein. This patient's TC level was >9.0 mmol/L. The pathogenicity of a leucine-to-proline mutation at the same location caused by a T>C base change was described by Sun et al.,[15]using cell extracts from Chinese FH patients. As shown in supplementary Fig. 2, the leucine residue at this position is highly conserved across several species, and PolyPhen, SIFT and MutationTaster predicted the variation to be 'damaging'.
The second novel variant, c.1187-25C>T, was the only identified variant in patient 6, a coloured patient of Tswana origin. The subject was 38 years old at the time of DNA blood draw with a TC level of 10.6 mmol/L, LDL-C 8.1 mmol/L and clinical signs of arcus and tendon xanthoma. Although this variant is located 25 bp from the start of exon 9, it is predicted to have a proximity effect on the 3' acceptor splice (AG) consensus sequence by Human Splicing Finder (HSF) algorithms, and to introduce a score variation of -59.6% between the wild-type and mutant motif (supplementary Fig. 3A). This score variation, which is well under the -30% for MaxEnt variation threshold, indicates that the mutation breaks the splice site based on literature datasets.[16] The estimated consensus value of the mutant sequence differed from the reference wild-type sequence by a significant 25% (variation threshold 10%), resulting in a predictable break in wild-type branch point due to the mutation and a potential alteration of splicing (supplementary Fig. 3B).
Regarding the APOB gene, a silent C-to-T base change at nucleotide position 10701 of codon 3567 p.(T3567T) in exon 26 of the APOB gene was found in patients 8, 11 and 16, all of whom manifested clinical signs of corneal arcus and two of whom had tendon xanthomas, and who were of Swazi, Tswana/Sotho and Tswana origin, respectively (Table 1). This synonymous variant is presented as a rare polymorphism on the exome variant database with T/C minor allele frequencies of 0.001 in Europeans and 0.07 in black LDL-C 19.0 mmol/L. Two of these subjects had clinical HoFH, and while it seems unlikely that this variant caused FH in these subjects, samples of relatives were not available to carry out co-segregation analysis of this variant and plasma lipid levels.
The causative mutations were only found in one of the three patients with a clinical diagnosis suggestive of HoFH. To confirm the sensitivity of the HRM method, FH patients of Afrikaner, Indian and different European ancestry were also analysed (supplementary Tables 1 - 3). Overall nine different point mutations and three different gross deletions in the LDLR gene were found, including two of the founder mutations previously reported in the SA population, namely p.(D227E) and p.(V429M), and one reported in subjects from the Indian subcontinent, p.(P685L). The common APOB mutation p.(R3527Q) was found in one patient.
Discussion
The black African SA patients screened in this study were from nine tribal groups, with a comparison group of non-African subjects of Caucasian-Afrikaner, Caucasian-European and Indian ancestry. In the 16 black African subjects, eight predicted FH-causing mutations were identified. Several mutations previously reported in black African subjects were seen,[11] and two novel variants were identified. Both novel variants were predicted to be pathogenic by in silico analysis, but while co-segregation studies in the relatives of these patients would be valuable, family members were unavailable for such studies.
The relatively low detection rate raises concern about the sensitivity of the HRM method used. While we have previously demonstrated that HRM is a sensitive and specific technique[13] for mutation detection in FH patients, and with detection of a wide range of LDLR point mutations and gross deletions in the comparison group examined, we acknowledge that next-generation sequencing (NGS) is now the preferred method of choice for molecular diagnosis. This work was commenced in 2013 when such methods were not widely available, and NGS remains an expensive technique and beyond the funds available for this research project. Also, while we screened the regions of the PCSK9 and APOB genes known to contain pathogenic mutations in subjects of European ancestry,[13] it is possible that a proportion of patients may have mutations in the exons not covered here. While pathogenic variants in other regions of APOB have been reported,[17] these are very rare. Similarly, gain-of-function FH-causing mutations in PCSK9 are also rare, for example accounting for <2% of FH patients in the UK. While loss-of-function variants have been reported in African American individuals (with low cholesterol levels), no gain-of-function variants have been reported in this population group as far as we are aware. We therefore estimate that complete coverage of these two genes may at best have increased the overall detection rate by 2 - 3%. Failure to identify the causative mutation in over half of the patients and in two-thirds with a clinical presentation of HoFH is therefore surprising, and may suggest that in these subjects the FH phenotype may be caused by novel loci, for example in the STAP1 gene,[18] which was not examined here.
It has recently been reported that in a considerable proportion of European subjects with a clinical diagnosis of FH for which no causative mutation can be found, a polygenic cause is the most likely explanation for their elevated LDL-C levels.[19] While this is a possible explanation for the cause of the FH phenotype in a proportion of our no-mutation patients, unfortunately the single-nucleotide polymorphisms used in the LDL-Gene score[19] in European subjects have not been validated for use in individuals of African origin, and owing to frequency differences and differences in linkage disequilibrium, it is likely that a different set of single-nucleotide polymorphisms would be needed.
The identification of these seven different FH-causing variants in our black African subjects underlines the heterogeneity of the cases of the FH phenotype in these subjects and demonstrates that comprehensive sequencing of the known FH-changing genes will be required to enable the efficient identification of patients with a monogenic cause of their phenotype for cascade testing of their relatives, so that early intervention with lipid-lowering statin therapy can be offered.
Acknowledgements. SEH is a British Heart Foundation Professor and is funded by a programme grant from the British Heart Foundation (PG08/008) and by the University College London National Institute for Health Research Hospitals Biomedical Research Centre. FR was funded by the SA National Research Foundation.
REFERENCES
1. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler K, eds. The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. New York: McGraw-Hill, 2001:2863-2914. [ Links ]
2. Marks D, Thorogood M, Neil HAW, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168(1):1-14. http://dx.doi.org/10.1016/S0021-9150(02)00330-1 [ Links ]
3. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: A HuGE prevalence review. Am J Epidemiol 2004;160(5):407-420. http://dx.doi.org/10.1093/aje/kwh236 [ Links ]
4. Santos RD, Gidding SS, Hegele RA, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: A consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol 2016;4(10):850-861. http://dx.doi.org/10.1016/S2213-8587(16)30041-9 [ Links ]
5. Usifo E, Leigh SEA, Whittall RA, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: Update and pathological assessment. Ann Hum Genet 2012;76(5):387-401. http://dx.doi.org/10.1111/j.1469-1809.2012.00724.x [ Links ]
6. Humphries SE, Whittall RA, Hubbart CS, et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: Relation to plasma lipid levels and coronary heart disease risk. J Med Genet 2006;43(12):943-949. http://dx.doi.org/10.1136/jmg.2006.038356 [ Links ]
7. Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A 2005;102(6):2069-2074. http://dx.doi.org/10.1073/pnas.0409736102 [ Links ]
8. Leren TP. Mutations in the PCSK9 gene in Norwegian subjects with autosomal dominant hypercholesterolemia. Clin Genet 2004;65(5):419-422. http://dx.doi.org/10.1111/j.0009-9163.2004.0238.x [ Links ]
9. Timms KM, Wagner S, Samuels ME, et al. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet 2004;114(4):349-353. http://dx.doi.org/10.1007/s00439-003-1071-9 [ Links ]
10. McPherson R. Monogenic hypercholesterolaemia. In: Dzau VJ, Liew C-C, eds. Cardiovascular Genetics and Genomics for the Cardiologist. Oxford: Blackwell Publishing, 2007:19-29. http://dx.doi.org/10.1002/9780470691977 [ Links ]
11. Loubser O, Marais AD, Kotze MJ, et al. Founder mutations in the LDL receptor gene contribute significantly to the familial hypercholesterolemia phenotype in the indigenous South African population of mixed ancestry. Clin Genet 1999;55(5):340-345. http://dx.doi.org/10.1034/j.1399-0004.1999.550507.x [ Links ]
12. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16(3):1215. http://dx.doi.org/10.1093/nar/16.3.1215 [ Links ]
13. Whittall RA, Scartezini M, Li K, et al. Development of a high-resolution melting method for mutation detection in familial hypercholesterolaemia patients. Ann Clin Biochem 2010;47(1):44-55. http://dx.doi.org/10.1258/acb.2009.009076 [ Links ]
14. Leitersdorf E, Hobbs HH, Fourie AM, Jacobs M, Westhuyzen DRV, Coetzee GA. Deletion in the first cysteine-rich repeat of low density lipoprotein receptor impairs its transport but not lipoprotein binding in fibroblasts from a subject with familial hypercholesterolemia. Proc Natl Acad Sci U S A 1988;85(21):7912-7916. http://dx.doi.org/10.1073/pnas.85.21.7912 [ Links ]
15. Sun XM, Patel DD, Webb JC, et al. Familial hypercholesterolemia in China: Identification of mutations in the LDL-receptor gene that result in a receptor-negative phenotype. Arterioscler Thromb Vasc Biol 1994;14(1):85-94. http://dx.doi.org/10.1161/01.ATV.14.1.85 [ Links ]
16. Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37(9):e67. http://dx.doi.org/10.1093/nar/gkp215 [ Links ]
17. Thomas ERA, Atanur SS, Norsworthy PJ, et al. Identification and biochemical analysis of a novel APOB mutation that causes autosomal dominant hypercholesterolemia. Mol Genet Genomic Med 2013;1(3):155-161. http://dx.doi.org/10.1002/mgg3.17 [ Links ]
18. Fouchier SW, Dallinga-Thie GM, Meijers JCM, et al. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ Res 2014;115(6):552-555. http://dx.doi.org/10.1161/CIRCRESAHA.115.304660 [ Links ]
19. Talmud PJ, Shah S, Whittall RA, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-controlstudy. Lancet 2013;381(9874):1293-1301. http://dx.doi.org/10.1016/S0140-6736(12)62127-8 [ Links ]
 Correspondence:
Correspondence:
S E Humphries
steve.humphries@ucl.ac.uk
Accepted 17 October 2016


{kind=link}
{kind=link}
{kind=link}