










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.107 no.2 Pretoria feb. 2017
http://dx.doi.org/10.7196/samj.2017.v107i2.12223
CME
Anaemia: Approach to diagnosis (part 2)
N AlliI; J VaughanII; M PatelIII
IMB BCh, FCPathHaem (SA); Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
IIMB BCh, FCPathHaem (SA), MMed (Haem); Department of Molecular Medicine and Haematology, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, and National Health Laboratory Service, Johannesburg, South Africa
IIIMB ChB, FCP (SA), MMed, FRCP (Lond), PhD; Department of Clinical Haematology, Division of Internal Medicine, Chris Hani Baragwanath Academic Hospital, Johannesburg, and School of Medicine, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Anaemia is defined as a condition in which the number of red cells or their oxygen-carrying capacity is insufficient to meet physiological needs. It is the most common disorder globally and one of the conditions that general practitioners most frequently encounter. In the World Health Organization global database, anaemia is estimated to affect 1.6 billion people. Anaemia may result from (i) decreased bone marrow output; or (ii) peripheral loss, destruction or sequestration of red cells. As anaemia manifests in a whole range of conditions, it is important to embrace a structured diagnostic approach. The recommended approach incorporates clinical and pathophysiological considerations, red cell characteristics, and bone marrow activity. Causes of anaemia related to decreased bone marrow output have been discussed in the previous issue of SAMJ, in the first part of this two-part series. The focus of the current article is on peripheral causes of anaemia.
This article is the second of a two-part CME series on anaemia.
According to the World Health Organization (WHO), anaemia is defined as a condition in which the number of red blood cells or their oxygen-carrying capacity is insufficient to meet physiological needs, which vary by age, sex, altitude, smoking, and pregnancy status.[1]
The causes of anaemia are divided into two broad categories (Fig. 1):
• Central: decreased bone marrow production/output of red blood cells, discussed in part 1 of the CME series on anaemia.[2]
• Peripheral: loss or destruction of red cells through haemolysis, bleeding or splenic sequestration, which is the subject of discussion in this issue.
While the two categories form a sound basis for diagnostic work-up, they are not mutually exclusive. As a rule, a good reticu-locyte response essentially excludes bone marrow failure. However, a combination of the two categories may give mixed signals that could present a confusing picture. By way of example, uncomplicated haemolysis with normal bone marrow function invokes a brisk reticulocyte response in the initial stages (Fig. 1), but with time, as folate is lost and stores are depleted, the response is blunted owing to ineffective haematopoiesis.
Haemolysis
Haemolysis is defined as the premature destruction of red cells. The lifespan of normal red cells is 100 - 120 days.
Haemolysis is classified as:
• intravascular, where red cells are destroyed within the vascular compartment
• extravascular, where red cells are destroyed outside the vascular system by macrophages.
It is important to understand the patho-physiology of the two categories of haemolysis, as clinical manifestations and treatment principles differ in the two types (Table 1).
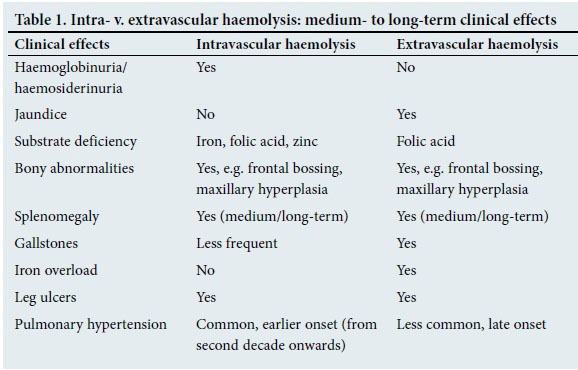
Intravascular haemolysis
Red cells break up within the vascular compartment and its contents are released into the circulating plasma. Free Hb tetramers dissociate into α/non-α dimers, which are small enough to filter through the glome-rulus and pass out into the urine (haemoglobinuria). Hb loss is accompanied by iron loss and may cause iron deficiency. Some of the filtered Hb is reabsorbed by the proximal tubular cell, where it is degraded, and the iron is packaged as haemosiderin. Tubular cells have a rapid turnover and these slough off into the urine. Haemosiderinuria is detected by microscopic viewing of a slide prepared from urine sediment. Iron present in tubular cells stains deep blue with the prussian blue iron stain.
Folic acid and carbonic anhydrase molecules are small enough to filter through the glomerulus and pass out into urine. A third of total body folic acid is stored in red cells; therefore, ongoing haemolysis is likely to cause megaloblastic anaemia secondary to folate loss and deficiency. Similarly, loss of carbonic anhydrase, a metallo-enzyme that contains zinc, could lead to zinc deficiency.
The red cell membrane comprises a large meshwork of interconnected lipopro-teins, and is therefore unable to be filtered through the kidneys. Massive intravascular haemolysis (IVH) (e.g. immediate blood transfusion reaction) frequently results in obstruction of glomerular capillaries and may cause pre-renal renal failure.
Extravascular haemolysis
Red cells are haemolysed within the macro-phage and its contents released into the cyto-sol. The haem component of Hb is enzymati-cally transformed to bilirubin, which is then released into the circulation. As unconjugated bilirubin is water insoluble, it is bound to albumin and the complex is finally absorbed by hepatocytes, where it is conjugated and excreted through the biliary system.
As there is no mechanism to release excess iron, it continues to accumulate over the duration of haemolysis. Folic acid is also released into the circulation, but much of it is lost through the kidneys.
Clinical manifestations and complications of chronic IVH and extravascular haemolysis (EVH) are summarised in Table 1.
It should be noted that haemolysis is seldom purely intravascular or extravascular. In instances where both IVH and EVH exist, the clinical picture is determined by the predominant type of haemolysis. Examples of conditions that cause haemolysis are summarised in Fig. 2.
Laboratory tests for haemolysis
The most sensitive laboratory indicator of haemolysis is a decreased haptoglobin level. Available tests for haemolysis are sum marised in Table 2.

Diagnostic approach to haemolysis
Clinical approach
A detailed history is essential, including duration and frequency of symptoms, age of onset, blood transfusions and family history. Inherited varieties generally present during early childhood.
Patients with homozygous beta thalassae-mia become progressively anaemic during the first few months of infancy and present at the age of ~3 - 6 months with severe anaemia. During the third trimester of pregnancy (~27 - 30 weeks), a switch from Hb F to Hb A production begins. Owing to non-functioning beta globin genes, there is no beta globin chain expression and therefore no Hb A production to substitute the gradual decline of Hb F. This results in a gradual but progressive drop in Hb during the first few months of infancy.
Patients with sickle cell disease (SCD) have a unique mode of presentation. The hallmark of SCD is an acute pain crisis, most commonly affecting long bones, although any site/organ may be affected. Severe pain is experienced at the site of vaso-occlusion. Dactylitis (pain crisis in the bones of the hands and feet) is often the presenting symptom of SCD in early childhood.[3]
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is inherited in an X-linked fashion and therefore typically manifests in males, although homozygous females are occasionally encountered. The haemolysis is most commonly episodic, following exposure to oxidant compounds, e.g. antimalarial drugs or sulphonamides. At birth, neonatal jaundice is common and may require intervention with ultraviolet light or exchange transfusion in more severe cases.
Of the red cell membrane disorders, hereditary spherocytosis is the most common variety. It presents with the classic triad of anaemia, jaundice and splenomegaly. The red cells are destroyed in the macrophages of the spleen, i.e. they undergo EVH.
In chronic haemolytic states, e.g. thalassae-mia major or intermedia, splenomegaly and bony abnormalities, such as frontal bossing, develop at an early age and are usually a sequel to inadequate management or poor patient compliance.[4]
Laboratory approach
A full blood count, differential count, reticu-locyte count and microscopic blood smear examination serve as baseline tests from whence further investigations are performed (Fig. 1). If haemolysis is suspected, it should be confirmed with a low haptoglobin level, which typically decreases during an haemo-lytic episode. Haptoglobin is a sensitive indicator of IVH and EVH. It should, however, be noted that haptoglobin levels may also decrease in liver disease (due to decreased synthesis) and rise in inflammatory states or malignancy (as it is an acute-phase reac-tant).[5] Peripheral blood smear microscopy (May-Grunwald-Giemsa and reticulocyte preparations) often provides the diagnosis or additional clues for further investigation (Table 3). Reticulocytes, Heinz bodies and Hb H inclusions in red cells can be detected through microscopic examination of smears prepared from reticulocyte preparations (peripheral blood incubated with brilliant cresyl blue for 20 minutes). Newer-generation blood cell counting analysers have the ability to enumerate reticulocytes.
Fragmentation haemolysis
Fragmentation haemolytic anaemias occur as a result of disturbances in blood flow, either in large (macroangiopathic haemolysis) or small (microangiopathic haemolysis) blood vessels. They are characterised by the presence of varying numbers of red cell fragments in the peripheral blood and accompanying thrombocytopenia in micro-angiopathic haemolysis.
Causes of macroangiopathic haemolysis include prosthetic heart valves, arterio-venous malformations and aortic stenosis. The anaemia is typically not severe, and active management is usually not required.
Microangiopathic haemolysis is caused by small-vessel obstruction by microthrom-bi, the distribution of which determines the organ system predominantly involved. Causes of microangiopathic haemolytic anaemia (MAHA), their associations and the specific clinical and laboratory derangements seen with each entity are summarised in Table 4. Several are pregnancy associated, and can be difficult to differentiate in the peripartum setting.
Thrombotic thrombocytopenic purpura (TTP) is an important cause of MAHA in South Africa (SA), as it is relatively common in HIV-positive patients. It carries a high mortality in the event of a missed diagnosis (~90%),[6] but with appropriate management survival is excellent (~80%).[7] Other causes include autoimmune disease, a variety of drugs (including clopidogrel, ticlodipine and quinine) and congenital deficiency of ADAMTS13 (a metalloproteinase that cleaves high molecular weight Von Willebrand factor). HIV remains the most common cause in SA. TTP is characterised by a pentad of features: fragmentation haemolysis, thrombocytope-nia, fever, renal failure (usually not severe), and fluctuating neurological derangement (including confusion or psychiatric manifestations, seizures and focal signs). However, all five features are not invariably present, and a presumptive diagnosis of TTP is made when there is MAHA and thrombocytopenia, with exclusion of all other causes of a thrombotic microangiopathy (e.g. disseminated intravascular coagulation). In the developed world, much emphasis is placed on the importance of documenting ADAMTS13 levels in the diagnosis of TTP, where its activity is reportedly consistently low (<10%) in congenital and autoimmune-mediated TTP, but normal or more modestly reduced in other micro-angiopathies.[7] However, ADAMTS13 levels are not universally low in HIV-associated TTP, which places the use of this test in question in the SA context.[8] Furthermore, ADAMTS13 testing is not universally available in SA, and it is therefore not routinely recommended.
Although the gold standard of treatment for TTP is plasma exchange, large-volume fresh frozen plasma may suffice in some patients. Corticosteroids should also be initiated at diagnosis, and HIV-seropositive patients should receive concomitant combination antiretroviral therapy, irrespective of their CD4 count. The lactate dehydrogenase level and platelet count are used to monitor response to treatment. Importantly, TTP is a relative contraindication for platelet transfusion, as this may fuel the microthrombotic process. However, platelets should not be withheld in the event of significant bleeding. Patients with TTP should be referred to and managed by specialist haematologists.
Bleeding
Anaemia due to acute bleeding usually occurs after substantial blood loss and is most often encountered in patients with a bleeding diathesis, or in trauma, postoperative or obstetric settings. In the hyper-acute phase of bleeding, the anaemia is usually normocytic and normochromic. This is followed by macrocytosis due to a reticulocyte response that develops over 2 - 3 days. Chronic low-volume blood loss causes a microcytic anaemia without an elevation of the reticulocyte production index (RPI) (due to iron deficiency). Where the source of the blood loss is not obvious, the following investigations may be helpful: (i) stool analysis for faecal occult blood; (ii) urine dipstix and microscopy for haemoglobinuria and detection of red cells, respectively; (iii) gynaecological review; and (iv) examination of sputum for iron-laden macrophages (detected in pulmonary haemosiderosis due to intra-alveolar bleeding).
Hypersplenism
Hypersplenism refers to an enlarged spleen with sequestration or destruction of blood cells, which leads to the development of anaemia and/or neutropenia and/or thrombocytopenia. It is characterised by a hypercellular bone marrow response and improvement after splenec-tomy. There may be some degree of associated haemolysis.
The spleen functions as a reservoir for blood cells and a filter that removes senescent, abnormal or antibody-coated red cells from the circulation. When it is enlarged, these functions are amplified; therefore, red cell survival is somewhat attenuated and the number of cells sequestered is increased. This may result in mild to moderate reductions in red cell, white cell and/or platelet counts, as well as biochemical evidence of haemolysis.
Hypersplenism may occur in any patient with splenomegaly, regardless of the cause, including portal hypertension, infectious causes (such as chronic malaria), chronic haemolytic states, or haematolo-gical malignancies. A peripheral blood smear review is routinely indicated in patients with splenomegaly, and bone marrow examination in instances where the cause for the splenomegaly is not readily apparent.
Where anaemia is caused by hypersplenism alone, i.e. without an underlying cause, the red cells are usually normocytic and normo-chromic. The reticulocyte count and RPI are usually raised, but may be lower than anticipated owing to sequestration of reticulocytes in the spleen. In patients with portal hypertension, anaemia is often microcytic and hypochromic because of iron deficiency from chronic blood loss, e.g. bleeding oesophageal varices, haematuria or gastrointestinal bleeding in chronic schistosomiasis. The latter is not an uncommon cause of portal hypertension in SA.
Conclusion
Causes of anaemia are numerous and varied. Owing to its manifestation in a wide variety of conditions, investigating the cause requires a thorough history, physical examination and systematic laboratory and ancillary investigations.
REFERENCES
1. World Health Organization. The Global Prevalence of Anaemia in 2011. Geneva: WHO, 2015. [ Links ]
2. Alli N, Vaughan J, Patel M. Anaemia: Approach to diagnosis. S Afr Med J 2017;107(1):23-27. http://dx.doi.org/10.7196/SAMJ.2017.v107i1.12147 [ Links ]
3. Alli NA, Patel M, Alli HA, et al. Recommendations for the management of sickle cell disease in South Africa. S Afr Med J 2014;104(11):743-751. http://dx.doi.org/10.7196/SAMJ.8470 [ Links ]
4. Olivieri NF, Weatherall DJ. Clinical aspects of β thalassaemia. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, eds. Disorders of Hemoglobin, Genetics, Pathophysiology and Clinical Management. Cambridge: Cambridge University Press, 2001. [ Links ]
5. Means RT Jr, Glader B. Anaemia: General considerations. In: Greer JP, Arbor DE, Glader B, et al., eds. Wintrobe's Clinical Hematology. 13th ed. Philadelphia: Wolters Kluwer, 2013. [ Links ]
6. Amorosi EL, Ultmann JE. Thrombotic thrombocytopenic purpura: Report of 16 cases and review of the literature. Medicine 1966;45(2):139-159. http://dx.doi.org/10.1097/00005792-196603000-00003 [ Links ]
7. Scully M, Goodship T. How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol 2014;164(6):759-766. http://dx.doi.org/10.1111/bjh.12718 [ Links ]
8. Gunther K, Garizio D, Nesara P. ADAMTS13 activity and the presence of acquired inhibitors in human immunodeficiency virus-related thrombotic thrombocytopenic purpura. Transfusion 2007;47(9):1710-1716. http://dx.doi.org/10.1111/j.1537-2995.2007.01346.X [ Links ]
 Correspondence:
Correspondence:
N Alli
nazeer.alli@nhls.ac.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}