










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.107 no.1 Pretoria ene. 2017
http://dx.doi.org/10.7196/samj.2017.v107i1.10907
RESEARCH
A novel FKRP-related muscular dystrophy founder mutation in South African Afrikaner patients with a phenotype suggestive of a dystrophinopathy
M M MudauI; F EssopII; A KrauseIII
IMSc (Biotech); Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIMSc (Med); Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIMB BCh, PhD; Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND. Fukutin-related protein (FKRP) muscular dystrophy is an autosomal recessive disorder caused by mutations in the FKRP gene. The condition is often misdiagnosed as a dystrophinopathy. A previously unreported mutation, c.1100T>C in exon 4 of FKRP, had been identified in homozygous form in two white South African (SA) Afrikaner patients clinically diagnosed with a dystrophinopathy. Objectives. To investigate whether the c.1100T>C mutation and the common European FKRP mutation c.826C>A are present in other patients of Afrikaner origin with suspected dystrophinopathy, and whether a founder haplotype exists.
METHODS. The c.1100T>C mutation was initially tested for using an amplification refractory mutation system technique in 45 white SA Afrikaner patients who had tested negative using multiplex ligation probe amplification screening for exonic deletions/duplications in the dystrophin gene. Sequencing analysis was used to confirm the c.1100T>C mutation and screen for the c.826C>A mutation. Two cohorts (each numbering 100) of Afrikaans and other white controls were screened for the c.1100T>C and c.826C>A mutations, respectively.
RESULTS. Of the 45 patients, 8 patients (17.8%) were homozygous for c.1100T>C, 2 (4.4%) were compound heterozygotes for c.1100T>C and c.826C>A, and 1(2.2%) was heterozygous for c.1100T>C with a second unidentified mutation. The c.1100T>C mutation was found in 1/100 controls, but no heterozygotes for the c.826C>A mutation were identified. Linked marker analysis for c.1100T>C showed a common haplotype, suggesting a probable founder mutation in the SA Afrikaner population.
CONCLUSION. FKRP mutations may be relatively common in Afrikaners, and screening should be considered in patients who have a suggestive phenotype and test negative for a dystrophinopathy. This test will be useful for offering diagnostic, carrier and prenatal testing for affected individuals and their families. As FKRP muscular dystrophy is autosomal recessive in inheritance, the implications of a positive diagnosis in a family differ significantly from those of an X-linked dystrophinopathy.
Fukutin-related protein (FKRP; OMIM #606596) muscular dystrophies are autosomal recessive disorders caused by mutations that occur in the FKRP gene. They may be misdiagnosed as dystrophinopathies because they have overlapping clinical features. Mutations in the coding exon of FKRP cause both merosin-deficient congenital muscular dystrophy type 1C (MDC1C) and limb-girdle muscular dystrophy type 2I (LGMD2I).[1-3] MDC1C represents a more severe phenotype, while LGMD2I is the milder allelic version.[1] Duchenne's muscular dystrophy (DMD; OMIM #310200) and Becker's muscular dystrophy (BMD; OMIM #300376), grouped together as dystrophinopathies, are both X-linked recessive disorders involving the proximal skeletal muscles. Dystrophinopathies are caused by mutations in the dystrophin gene, leading to complete inactivation (DMD) or partial inactivation (BMD) of this gene.[4]
The FKRP gene maps to the cytogenetic region 19q13.32 between the markers D19S219 and D19S606. It is composed of four exons and spans 12.53 kb. Exon 4, which is the largest and the only coding exon in this gene, contains a portion of the 5' untranslated region (UTR), as well as the entire protein coding region and the complete 3' UTR. The FKRP gene encodes a 495 amino acid protein known as the fukutin-related protein (FKRP).1
Founder mutations occur in populations that originate from a small number of individuals and undergo a period of rapid growth. The Afrikaner population of South Africa (SA) has many features of a founder population. The term 'Afrikaners' refers to a subset of white SA individuals who are descended from a small group of German, Dutch and Huguenot settlers who immigrated to the Cape of Good Hope in the 1600s.[6] It is difficult to distinguish Afrikaner individuals from other South Africans of European ancestry, and usually surnames, as well as the use of Afrikaans as a first language, are used to broadly define an Afrikaner individual. There are many diseases for which founder mutations have been described in the Afrikaner population. Some of these include Fanconi's anaemia, porphyria variegata, familial hypercholesterolaemia, Gaucher's disease and autosomal recessive polycystic kidney disease.[7-11]
FKRP founder mutations have been described in a number of populations around the world, for example the c.826C>A (p.Leu276Ile) FKRP mutation in 20 LGMD German patients[12] as well as 13 LGMD patients from Denmark,[13] and a novel homozygous missense mutation, c.1364C>A (p.Ala455Asp), in Tunisian families.[14] A previously unreported mutation, c.1100T>C (p.Ile367Thr), was found in homozygous form in the FKRP gene in two unrelated SA white patients of Afrikaner origin clinically diagnosed with a dystrophinopathy in 2010 by the Genetics Centre at Guy's Hospital in London, and a founder mutation was therefore suspected.
Objectives
To screen for the c.1100T>C and the common European c.826C>A FKRP-related muscular dystrophy mutations in white SA Afrikaner patients who were referred for dystrophinopathy testing but tested negative for deletions or duplications in the dystrophin gene. The specific objectives were to determine whether SA Afrikaner patients clinically diagnosed with a phenotype suggestive of a dystrophinopathy and negative for dystrophin mutations have the c.1100T>C and c.826C>A FKRP mutations, and whether the c.1100T>C mutation is a founder mutation in the SA Afrikaner population based on the presence of a common haplotype.
Subjects
Patients
A total of 45 white SA patients of Afrikaner origin (based on their surnames) were included in this study. The patients were routine referrals to the Division of Human Genetics, National Health Laboratory Service (NHLS), for dystrophinopathy diagnostic testing from 1994 to 2013 and who tested negative for exonic deletions/ duplications on multiplex ligation probe amplification (MLPA) screening using SALSA MLPA kits and P034 and P035 probe mixes (MRC-Holland, Netherlands). Patients with a family history suggestive of an X-linked pattern of disease were excluded.
The majority of referrals were from the state hospitals in Johannesburg (Charlotte Maxeke Johannesburg Academic Hospital, Chris Hani Baragwanath Hospital, Rahima Moosa Mother and Child Hospital) and from medical geneticists at the NHLS genetic counselling clinics. A small number of cases were referred from other genetic centres around SA and from doctors in private practice. Blood samples from the patients were sent in with a referral form stating the possible diagnosis and in some cases the clinical features observed.
Controls
A total of 200 unrelated and unaffected randomly selected white individuals whose DNA samples were taken from the DNA bank in the Division of Human Genetics were screened. Of these,100 were selected based on their Afrikaner surnames and screened for the c.1100T>C mutation, and a further 100 white controls were screened for the c.826C>A FKRP mutation.
Methods
All 45 patients were screened for the c.1100T>C mutation using the amplification refractory mutation system-polymerase chain reaction (ARMS-PCR) method, and positive results were confirmed on Sanger sequencing. The c.826C>A mutation was tested using Sanger sequencing only.
Techniques used
ARMS-PCR for FKRP (c.1100T>C) mutation
A multiplex ARMS-PCR method was designed for detection of the c.1100T>C mutation using four primers: primers FKRP Ex4-Control (F-outer) 5,-CGCCCGCCTACCTCTACGAG-3, and FKRP-ARMS-MUTANT (R-inner) 5,-GCCCACGTCCTCCAAGTTGG-3',which amplified the mutant sequence (209 base pairs (bp)), and primers FKRP-ARMS-NORMAL (F-inner) 5'-GGACTACGACGTGGACCTGGGTAT-3' and FKRP Ex4-Control (R-outer) 5'-AGA AGG GCC ACA GGT CCA CG-3', which amplified the normal sequence (186 bp). An internal control fragment of 351 bp was amplified using the FKRP Ex4-Control (F-outer) and FKRP Ex4-Control (R-outer) primers. The PCR products were separated on a 3% agarose gel.
Sequencing
Sanger sequencing was performed to confirm the presence of the two FKRP mutations. Products were run on a ABI 3130xl genetic analyser (Applied Biosystems, SA) and results were analysed using the SeqMan II application, DNASTAR Lasergene programme (DNASTAR, USA).
c.1100T>C mutation. The FKRP Ex4-Control primers (F-outer and R-outer) were used to confirm the ARMS-PCR results on patients who tested positive for the c.1100T>C mutation.
c.826C>A mutation. Published primers 3F [5,-ACC AGC CTC TTT CTG CAG ACC-3,] and 4R [5,-CCT TCT CCC ATA CGA AGC-3,][12] were used to amplify and sequence a 532-bp PCR fragment containing the c.826C>A FKRP mutation.
Haplotype analysis
Three dinucleotide short tandem repeat (STR) microsatellite markers flanking the FKRP gene, D19S219, D19S412 and D19S606, were typed in affected individuals and their families (where available) to determine whether the c.1100T>C mutation occurs on a founder haplotype.[5] The order of markers is centromere-D19S219-D19S412-FKRP-D19S606-telomere. The distance between D19S219 and D19S606 is ~1.9 MB, with ~0.72 MB between D19S606 and the mutation.[1]
Ethics approval
The project protocol was submitted to the Human Research Ethics Committee (Medical) of the University of the Witwatersrand, Johannesburg, and an ethical clearance certificate was obtained (ref. no. M120618)
Results
c.1100T>C and c.826C>A mutation screening
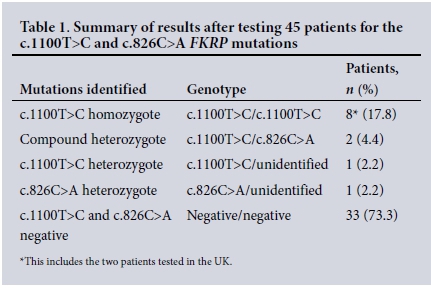
The diagnosis of FKRP muscular dystrophy was confirmed in 10/45 patients tested (22.2%) and was suggestive in another 2/45 heterozygous patients (4.4%) (Table 1).One heterozygous individual with the c.1100T>C mutation was identified in the 100 Afrikaner controls tested. None of the 100 white controls tested positive for the c.826C>A mutation.
Haplotype analysis
Three flanking dinucleotide STR markers, D19S219, D19S412 and D19S606, were used to determine a haplotype, with alleles 182 bp, 103 bp and 174 bp, respectively, tracking with the c.1100T>C mutation. Statistical analysis using Fisher's exact test showed that there were highly significant differences when patients were compared with normal controls between the frequencies of alleles 182 bp,103 bp and 174 bp, with p-values of <0.0001 for alleles 182 bp and 103 bp and p=0.0069 for allele 174 bp. This finding strongly suggests that a founder haplotype with alleles 182 bp, 103 bp and 174 bp exists in this population on the chromosome carrying the c.1100T>C mutation.
Discussion
The FKRP-related muscular dystrophies are caused by different mutations in the FKRP gene and may vary in disease severity.[2] The marked clinical similarity between LGMD2I and dystrophinopathy patients has often resulted in misdiagnosis of an FKRP-related muscular dystrophy.[3]
A study by Schwartz et a/.[13] in Denmark involved screening for the common c.826C>A (p.Leu276Ile) FKRP mutation in 102 European patients, who presented with a dystrophinopathy-like phenotype but in whom no dystrophin gene rearrangements were identified. The study found that ~13% of patients were homozygous for a common c.826C>A FKRP mutation and were therefore diagnosed with LGMD2I (an FKRP-related muscular dystrophy) and not with a dystrophinopathy as had initially been thought.
Similarly, before this study, a novel c.1100T>C (p.Ile367Thr) FKRP mutation was first identified in a homozygous state in two unrelated Afrikaner patients who were clinically diagnosed with DMD/BMD, but in whom no deletions or duplications in the dystrophin gene were found. As a result of this finding, a further 43 SA white patients suspected of having a dystrophinopathy were screened for this novel mutation in this study. Eight patients (17.8%) in total were found to have this mutation in a homozygous state and 3 (4.4%) were heterozygous for the mutation, suggesting that FKRP mutations are a significant cause of disease in patients presenting with a dystrophinopathy-like phenotype. Furthermore, since a significant number of patients with Afrikaans ancestry carry the c.1100C>T mutation, this mutation has not been reported in other populations, and linked marker analysis revealed a common haplotype, it is probable that the c.1100C>T mutation is a founder mutation in the Afrikaner population. A number of founder mutations for other genetic disorders have been identified in the SA Afrikaner population.[7-11]
Owing to the suggestive clinical features, the 3 heterozygous patients were subsequently screened for the c.826C>A mutation. This mutation was found in 2 of the 3, confirming the diagnosis of FKRP. muscular dystrophy. A diagnosis of FKRP was therefore confirmed in 22.2% of the patients screened and suspected in another 4.4% who were heterozygous for one of the mutations. It has been reported in one study[5] that 4 cases in 10 families (40%) were compound heterozygotes and carried another missense mutation in the second allele that was said to be pathogenic. In another study that screened for the common c.826C>A FKRP mutation in 20 German patients from 19 unrelated families, 13 of the 20 patients (65%) were found to be homozygous for the mutation and 3 were found to be compound heterozygotes carrying a second heteroallelic mutation.[12]
The FKRP c.826C>A mutation appears to be common worldwide, with a carrier frequency estimated to be 1 in 300.[12,15] In this current study, the carrier frequency of the c.1100T>C mutation was found to be approximately 1 in 100. This suggests that the FKRP c.1100T>C mutation may be more common in the Afrikaner population of SA than elsewhere. The c.826C>A mutation was not found in 100 control individuals tested in the SA population, but was found in the patient cohort, indicating that this is not a particularly common mutation in SA, but does occur.
Conclusion
This study shows that FKRP-related muscular dystrophy accounts for a significant number of patients with a phenotype suggestive of a dystrophinopathy, and that the mutation c.1100T>C is likely to be a founder mutation in the SA Afrikaner population. The c.826C>A was identified in compound heterozygous form, suggesting that this common European mutation also occurs in SA white patients. Patients of possible Afrikaans ancestry with a dystrophinopathy phenotype who test negative for DMD/BMD mutations on MLPA should be screened for FKRP mutations. The findings emphasise the need for caution in the diagnosis of dystrophinopathy-like disorders, to prevent misdiagnosis and inaccurate genetic counselling. Since FKRP-related muscular dystrophy is an autosomal recessive and not an X-linked disorder, the risks to relatives are significantly different. The knowledge derived from this study will be very useful in ensuring more accurate diagnostic, carrier and prenatal testing for SA families with dystrophinopathy-like muscular dystrophies.
Acknowledgements. The authors thank the NHLS for funding this project (NHLSRT grant no. 94308) and the Genetics Centre at Guy's Hospital, London, for the initial diagnosis.
REFERENCES
1.Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 2001;10(25):2851-2859. http://dx.doi.org/10.1093/hmg/10.25.2851 [ Links ]
2. Topaloglu H, Brockington M, Yuva Y, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 2003;60(6):988-992. http://dx.doi.org/10.1212/01.WNL.0000052996.14099.DC [ Links ]
3. Poppe M, Cree L, Bourke J, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003;60(8):1246-1251. http://dx.doi.org/10.1212/01.WNL.0000058902.88181.3D [ Links ]
4. Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am J Hum Genet 1989;45(4):498-506. [ Links ]
5. Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am J Hum Genet 2001;69(6): 1198-1209. http://dx.doi.org/10.1086/324412 [ Links ]
6. Botha MC, Beighton P. Inherited disorders in the Afrikaner population of southern Africa: Part II. Skeletal, dermal and haematological conditions; the Afrikaners of Gamkaskloof; demographic considerations. S Afr Med J 1983;64(17):664-667. [ Links ]
7. Tipping AJ, Pearson T, Morgan NV, et al. Molecular and genealogical evidence for a founder effect in Fanconi anemia families of the Afrikaner population of South Africa. Proc Natl Acad Sci U S A 2001;98(10):5734-5739. http://dx.doi.org/10.1073/pnas.091402398 [ Links ]
8. Meissner PN, Dailey TA, Hift RJ, et al. A R59W mutation in human protoporphyrinogen oxidase results in decreased enzyme activity and is prevalent in South Africans with variegate porphyria. Nat Genet 1996;13(1):95-97. http://dx.doi.org/10.1038/ng0596-95 [ Links ]
9. Callis M, Jansen S, Thiart R, de Villiers JN, Raal FJ, Kotze MJ. Mutation analysis in familial hypercholesterolemia patients of different ancestries: Identification of three novel LDLR gene mutations. Mol Cell Probes 1998;12(3):149-152. http://dx.doi.org/10.1006/mcpr.1998.0164 [ Links ]
10. Morar B, Lane AB. The molecular characterization of Gaucher disease in South Africa. Clin Genet 1996;50(2):78-84. http://dx.doi.org/10.1111/j.1399-0004.1996.tb02352.x [ Links ]
11. Lambie L, Amin R, Essop F, Cnaan A, Krause A, Guay-Woodford LM. Clinical and genetic characterization of a founder PKHD1 mutation in Afrikaners with ARPKD. Pediatr Nephrol 2015;30(2):273-279. http://dx.doi.org/10.1007/s00467-014-2917-1 [ Links ]
12. Walter MC, Petersen JA, Stucka R, et al. FKRP (826C>A) frequently causes limb-girdle muscular dystrophy in German patients. J Med Genet 2004;41(4):e50. http://dx.doi.org/10.1136/jmg.2003.013953 [ Links ]
13. Schwartz M, Hertz JM, Sveen ML, Vissing J. LGMD2I presenting with a characteristic Duchenne or Becker muscular dystrophy phenotype. Neurology 2005;64(9):1635-1637. http://dx.doi.org/10.1212/01.WNL.0000157654.59374.E5 [ Links ]
14. Louhichi N, Triki C, Quijano-Roy S, et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities: Identification of a founder mutation in Tunisian families. Neurogenetics 2004;5(1):27-34. http://dx.doi.org/10.1007/s10048-003-0165-9 [ Links ]
15. De Paula F, Vieira N, Starling A, et al. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: The other end of the spectrum. Eur J Hum Genet 2003;11(12):923-930. http://dx.doi.org/10.1038/sj.ejhg.5201066 [ Links ]
 Correspondence:
Correspondence:
M M Mudau
maria.mudau@nhls.ac.za
Accepted 9 September 2016.

