










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.106 n.3 Pretoria Mar. 2016
http://dx.doi.org/10.7196/samj.2016.v106i3.10005
RESEARCH
Alpha-thalassaemia trait as a cause of unexplained microcytosis in a South African population
S B LoonatI; N H NaranII; S L TheinIII; N A AlliIV
IMT (Haem), MSc; Department of Molecular Medicine and Haematology, School of Pathology, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
IIPhD; Department of Chemical Pathology, School of Pathology, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
IIIMBBS, FRCP, FRCPath, DSc; Department of Molecular Haematology, King's College London, UK
IVMB BCh, FCPath (Haem); Department of Molecular Medicine and Haematology, School of Pathology, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
ABSTRACT
BACKGROUND: Red cell microcytosis is a common abnormality detected in a full blood count, which often prompts clinicians to investigate further for a cause. In the absence of iron deficiency and anaemia of chronic disease, the differential diagnosis includes β-thalassaemia trait and α-thalassaemia trait.
METHODS: We investigated the contribution of α-thalassaemia trait in South African subjects with unexplained microcytosis. Iron studies, haemoglobin subfractionation and multiplex polymerase chain reaction (PCR) analysis for α-globin gene deletions were performed on 97 controls and 86 patients.
RESULTS: After excluding iron deficiency, anaemia of chronic disease and β-thalassaemia trait, 78.0% of subjects with unexplained microcytosis were confirmed on PCR analysis to have α-thalassaemia trait.
CONCLUSION: Alpha-thalassaemia trait accounts for the majority of unexplained microcytosis.
Microcytosis, with or without red cell hypochromia, is a common abnormality detected in a full blood count (FBC) and often prompts clinicians to investigate further for a cause. The differential diagnosis of microcytic and hypochromic red cell indices includes iron deficiency anaemia, anaemia of chronic disease, α- or β-thalassaemia trait, and sideroblastic anaemia. In the absence of iron deficiency and chronic disease, clinicians often request a haemoglobinopathy screen to exclude thalassaemia.
Whereas heterozygous β-thalassaemia is readily identified through an increased haemoglobin (Hb)A2 level using Hb separation techniques, without DNA analysis α-thalassaemia trait remains a diagnosis of exclusion. Although HbH inclusions are occasionally observed on incubated reticulocyte preparations in two-gene deletion α-thalassaemia, the majority of α-thalassaemia traits remain undetected through routine laboratory methods.
In a quest to distinguish iron deficiency from thalassaemia trait, several formulae utilising red cell indices from a routine FBC have been proposed.[1] These serve as good screening tools, but are unable to distinguish between α- and β-thalassaemia.
Since definitive exclusion of α-thalassaemia can only be obtained through molecular characterisation of the α-globin gene, the most logical diagnostic approach for unexplained microcytosis would be to use polymerase chain reaction (PCR) analysis to detect the common α-globin gene deletions. In this study we employed a multiplex PCR assay designed to detect the seven most common α-thalassaemia deletions.[2,3]
Methods
Patients and controls
Patient samples were selected from routine specimens received for FBC analysis at the Chris Hani Baragwanath Academic, Charlotte Maxeke Johannesburg Academic and Helen Joseph Hospital laboratories in Johannesburg, South Africa. A total of 100 patient samples were collected over a period of 36 months, of which 86 fulfilled the criteria for unexplained microcytosis, as defined below.
Inclusion criteria
Inclusion criteria were: (i) microcytosis with a mean cell volume (MCV) <81 fl; (ii) mean cell haemoglobin (MCH) <27 pg; (iii) normal/borderline serum iron studies (ferritin 30 - 400 μg/L (male), 15 - 150 μg/L (female), and serum iron 9 - 30 μmol/L); and (iv) normal/decreased HbA2 level (normal 2.4 - 3.7%).
Control samples (N=146) were obtained from volunteer staff members (aged >18 years). Those with normal FBCs and iron profiles were enlisted as controls. Among the 146 control subjects, 49 had microcytosis and were therefore excluded from the control group.
Analysis
The following tests were performed on the control and patient groups:
1. FBC and reticulocyte count (automated Sysmex XE 5000, analyser; Sysmex Incorporated, Japan), blood smears for microscopy (automated Sysmex 1000i slide maker and stainer; Sysmex Incorporated, Japan), and reticulocyte preparation for microscopy (2-hour incubation with 1% Brilliant Cresyl Blue supravital stain; National Health Laboratory Service, South Africa).
2. Serum iron studies including serum iron, serum ferritin, serum transferrin and percentage saturation (Cobas analyser; Sysmex Incorporated, Japan) using the ferrozine method without deproteinisation for serum iron, and immunological agglutination principle for transferrin and ferritin). The percentage saturation was calculated using the serum iron and transferrin levels.
3. Relative quantification of the Hb subtypes. This was obtained using cation exchange high-pressure liquid chromatography (HPLC) (Bio-Rad D10 analyser; Bio-Rad Laboratories, USA). All abnormal results on HPLC were confirmed with alkaline and acid pH Hb electrophoresis (HYDRASYS system, Sebia-Norcross; SEBIA-INC, USA)
4. Multiplex PCR was performed as described by Chong et al.[3] DNA was extracted using a High Pure PCR preparation kit (Roche Diagnostics, Germany).
Alpha-thalassaemia PCR methodology
On initial set-up the PCR reaction for each genotype was optimised individually (Table 1), and then adapted for all seven genotypes in a multiplex system with the respective sets of primers.
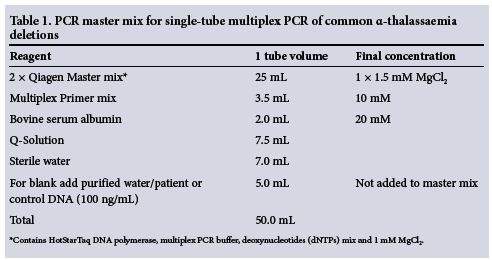
The volumes referred to in Table 1 are for a single PCR reaction. The conditions of the cycler were programmed as follows: PCR reaction initiated at 95°C for 10 minutes followed by 35 cycles, each comprising: (i) denaturation step -> 45 seconds at 97°C; (ii) annealing step -- 90 seconds at 61°C; and (iii) initial elongation step - 72°C for 90 seconds. A final elongation step then followed the 35 cycles at 72°C for 5 minutes.
The deletions were detected through separation of the PCR products by electrophoresis on a 1.5% agarose gel (stained with ethidium bromide). The bands were visualised and photographed under ultraviolet transillumination, and identified using known controls (complements of AS Tan, Department of Paediatrics, National University of Singapore, and SL Thein, King's College London Hospital, UK). A 1 kb DNA marker was included in each run (Figs 1 and 2).


Statistical analysis
Statistical analysis was carried out using the Statistical Analysis System (SAS) program, version 9.1 (North Carolina State University, USA). The Wilcoxon two-sample test was used to analyse the haematological and biochemical parameters between the patient and control groups. Fisher's exact test was used to calculate the frequency of the genotypes and the percentage differences in the population groups.
Results
All patients and control subjects were adults, with mean ages for the two groups of 34.5 (standard deviation (SD) 14.5) and 36.4 (SD 12.7) years, respectively. The gender split showed male/female ratios of 0.45 and 0.46 in the patient and control groups, respectively. Ethnic groups were determined as per hospital classification. In the patient group, blacks, Indians and whites comprised 51.1%, 40.7% and 8.1%, respectively. There were two subjects of mixed ancestry, who were incorporated into the black group for analysis. A similar distribution was noted in the control group, where blacks, Indians and whites comprised 54.6%, 28.8% and 16.4%, respectively.
Haematological parameters including Hb, MCV, MCH, RDW (red cell distribution width) and HbA2 were significantly lower in the patient group than in the control group (p<0.05) (Table 2).

Transferrin and ferritin levels showed no significant difference between the patient and control groups. Serum iron levels, however, displayed a statistically significant difference (p=0.0441) between patients and controls, but these values were within normal limits (>10 μmol/L).
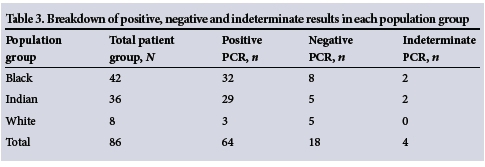
Multiplex PCR for detecting the seven common deletional α-thalassaemia variants was performed in all patient and control subjects (Figs 1 and 2). Of a total of 86 in the patient group, 64 (78.0%) tested positive for α-thalassaemia. The prevalences in the black, Indian and white population groups were 80.0%, 85.3% and 37.5%, respectively.
There were four indeterminate results, which were excluded from analysis. This left 18 subjects (22.0%) for whom the cause of the microcytosis remains unexplained (Table 3). Multiplex PCR analysis on control subjects yielded negative results.
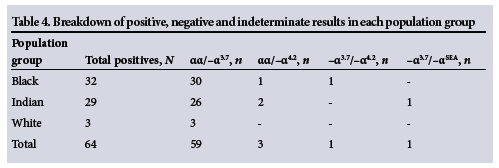
The breakdown of the various α-thalass-aemia genotypes encountered in the study group is detailed in Table 4. The most common α-thalassaemia genotype was -α3.7, which accounted for 92.1%, whereas the -α4.2 genotype accounted for 4.7% of the total number of positive results. In addition, two subjects had a double heterozygous genotype, viz.-a3,7/-a4,2 and -α3,7/--SEA.
The subject with the -α3.7/--SEA mutation was of Malaysian descent with HbH disease and was incorporated into the Indian group for analysis.
Hb separation revealed seven subjects with sickle cell disease, of whom five had α-thalassaemia on PCR.
Discussion
Red cell microcytosis and hypochromia are commonly encountered in the clinical setting. Differentiating between the causes of microcytosis (with or without hypochromia) poses a challenge to clinicians. Not only does an accurate diagnosis allow for appropriate management and counselling, it also has therapeutic implications, particularly with regard to unnecessary iron therapy. Iron deficiency and chronic disorder remain the leading causes of microcytosis, followed by α-thalassaemia variants caused by deletion of α-globin gene/s, which is the most frequent haemoglobinopathy globally.[4] More than 40 different deletional variants of α-thalassaemia have been described.[5] Globally, the majority of α-thalassaemia is caused by the seven deletions tested for in this study.[2]
Of 86 subjects in the patient group, 78.0% tested positive for α-thalassaemia PCR, which leaves 22.0% for whom the cause of the microcytosis remains unexplained. Possible reasons for a negative PCR result include: (i) the presence of α-thalassaemia deletions not tested for, such as the αα/--SA mutation,[6] and (ii) α-thalassaemia caused by non-deletional α-gene mutations, which are uncommon. Sequencing of α-globin genes would be necessary to detect non-deletional α-thalassaemia mutations, as these are not detected by Gap PCR.
The most common deletional variant in this study was the αα/-α3.7 genotype (92.1%). The prevalence of the αα/-α3.7 genotype was similar in the black and Indian groups, viz. 80.0% and 85.3%, respectively. Other α-thalasaemia genotypes were uncommon, with αα/-α4.2 and αα/--SEA constituting 4.7% and 1.6% (single case), respectively. The subject with the αα/--SEA deletion had HbH disease and HbH inclusions were detected on microscopy (incubated reticulocyte preparation) in >50% of the red cell population. This finding is not surprising as the patient was of Malaysian descent, a population group in which the deletion is found in high frequencies.[7] Double α-globin deletions in cis (α0) were not encountered in black subjects in our study group. This finding is not unexpected since α0 deletions are rare in the black population, which also explains the rarity of HbH disease in this ethnic group.[8-9]
This pattern of alpha genotype distribution has been mirrored in other studies. Sankar et al.[10] reported the incidence of -/α3.7 as the most common genotype (35.7%) in microcytic hypochromic subjects of Indian descent. In their study, other causes of microcytosis such as iron deficiency and β-thalassaemia trait were not excluded from the cohort. A Canadian group[11] yielded a PCR-positive rate of only 24.5% in their unexplained microcytosis cohort. In their study, only 29% of the population were of Asian descent (immigrants), with the remainder being of Canadian descent. Of the positive PCR results, αα/-α3.7, αα/-α4.2, aa/-aSEA and αα/-αMED comprised 81.25%, 6.25%, 10.4% and 2.0% of the total, respectively. In both the studies above, -α3.7 was the most common mutation as a cause for α-thalassaemia.
In our study, of the 49 subjects excluded from the control group, 26 were confirmed to have iron deficiency and the remaining 23 tested positive for α-globin gene deletion/s. In the unadjusted control population of 120 subjects (97 normal controls + 23 controls with α-thalassaemia on PCR analysis and excluded from the control group), the extrapolated incidence of α-thalassaemia in the normal population is calculated as 19.1%.
Two additional haemoglobin abnormalities were detected in the control group viz., heterozygous β-thalassaemia (in two subjects), and hereditary persistence of fetal haemoglin (HPFH) with an HbF level of 30% and pancellular distribution of HbF (in one subject).
In resource-poor settings, PCR analysis may prove prohibitive for routine testing. However, if the MCV were utilised as a screening tool, a significant percentage of cases would remain unsuspected and/or undetected, a scenario that is far from ideal, particularly for prenatal and premarital screening. Multiplex PCR would offer a cost-effective solution for the detection of α-globin gene deletions.
Conclusion
Unexplained microcytosis is a frequently encountered problem in clinical practice. In this study, α-globin gene deletions accounted for 78% of unexplained microcytosis. Twenty-two percent of cases remain unexplained, and further studies are warranted to explore the possibility of other genetic defects in the α-globin cluster.
References
1. Narchi H, Basak RB. Comparison of erythrocyte indices to differentiate between iron deficiency and alpha-thalassaemias in children with microcytosis and/or hypochromia. East Mediterr Health J 2010;16(9):966-971. [ Links ]
2. Tan AS, Quah TC, Low PS, Chong SS. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood 2001;98(1):250-251. [http://dx.doi.org/10.1182/blood.V98.1.250] [ Links ]
3. Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood 2000;95(1):360-362. [ Links ]
4. Mach-Pascual S, Darbellay R, Pilotto PA, Beris P. Investigation of microcytosis: A comprehensive approach. Eur J Haematol 1996;57(1):54-61. [http://dx.doi.org/10.1111/j.1600-0609.1996.tb00490.x] [ Links ]
5. Cao A, Rosatelli MC, Monni G, Galanello R. Screening for thalassemia: A model of success. Obstet Gynecol Clin North Am 2002;29(2):305-328,vi-vii. [http://dx.doi.org/10.1016/S0889-8545(01)00006-7] [ Links ]
6. Vandenplas S, Higgs DR, Nicholls RD, Bester AJ, Mathew CG. Characterization of a new alpha zero thalassaemia defect in the South African population. Br J Haematol 1987;66(4):539-542. [http://dx.doi.org/10.1111/j.1365-2141.1987.tb01341.x] [ Links ]
7. Kattamis AC, Camaschella C, Sivera P, Surrey S, Fortina P. Human alpha-thalassemia syndromes: Detection of molecular defects. Am J Hematol 1996;53(2):81-91. [http://dx.doi.org/10.1002/(SICI)1096-8652(199610)53:2<81::AID-AJH5>3.0.CO;2-#] [ Links ]
8. Higgs DR, Pressley L, Old JM, et al. Negro alpha-thalassaemia is caused by deletion of a single alpha-globin gene. Lancet 1979;2(8137):272-276. [http://dx.doi.org/10.1016/S0140-6736(79)90290-3] [ Links ]
9. Felice AE, Cleek MP, McKie K, McKie V, Huisman TH. The rare alpha-thalassemia-1 of blacks is a zeta alpha-thalassemia-1 associated with deletion of all alpha- and zeta-globin genes. Blood 1984;63(5):1253-1257. [ Links ]
10. Sankar VH, Arya V, Tewari D, Gupta UR, Pradhan M, Agarwal S. Genotyping of alpha-thalassemia in microcytic hypochromic anemia patients from North India. J Appl Genet 2006;47(4):391-395.[http://dx.doi.org/10.1007/BF03194650] [ Links ]
11. Bergeron J, Weng X, Robin L, Olney HJ, Soulieres D. Prevalence of alpha-globin gene deletions among patients with unexplained microcytosis in a North-American population. Hemoglobin 2005;29(1):51-60.[http://dx.doi.org/10.1081/HEM-47024] [ Links ]
 Correspondence:
Correspondence:
N A Alli
nazeer.alli@nhls.ac.za
Accepted 27 October 2015.

